Human Genetics
1/21/2025
What is a genome?
all genetic information contained in a cell
3gbp for a haploid cell (but we have 2 copies so really 6 GBP)
Males actually have 6.27 GBP
Females actually have 6.369 GBP
Because Y chromosome is shorter
Genes
Gene: Basic physical and functional unit of heredity
Has to do something (encodes for a protein or other product like tRNA or rRNA)
Knowing what parts of the genome are functional lets us understand genetic disease
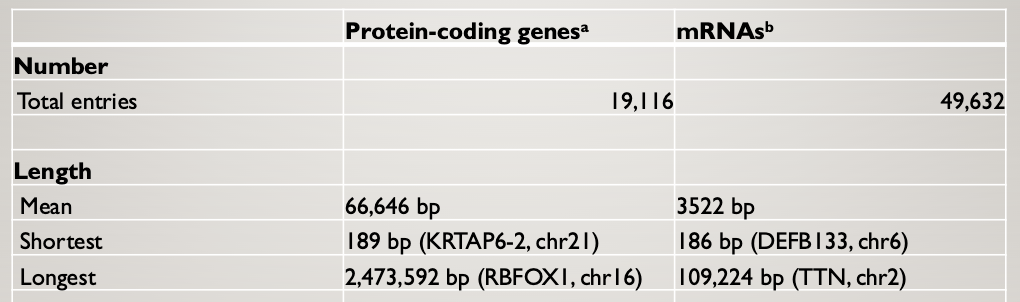
Humans have about the same number of PCGs as nematodes
Humans have many more mRNAs than protein-coding genes
Eukaryotic gene structure
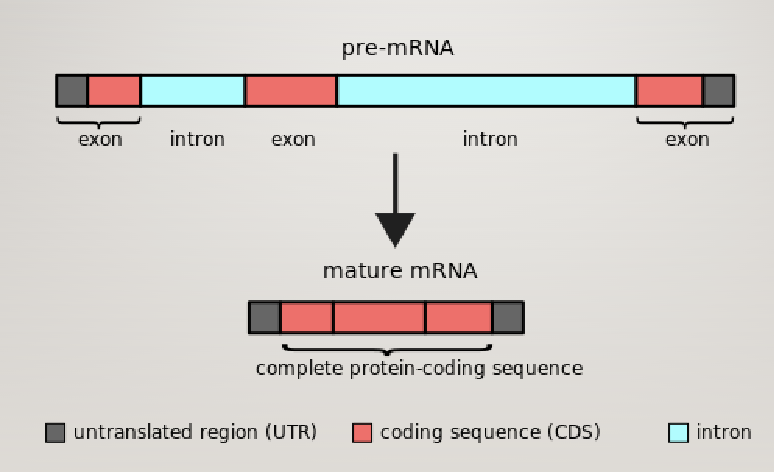
Pre-mRNA contains functional elements not present in mature mRNA
—Chromosomes
2 kinds (autosomes and sex chromosomes)
Distinguished because inherited diseases from sex chromosomes have different behaviors
We have 22 autosomes and X and Y sex chromosomes
Each autosome comes in two different versions in all your somatic cells — so 44
Plus XX or XY
Plus Mitochondrial
So we have 47 chromosomes
Chromosomes are numbered in order of length (longest to shortest)
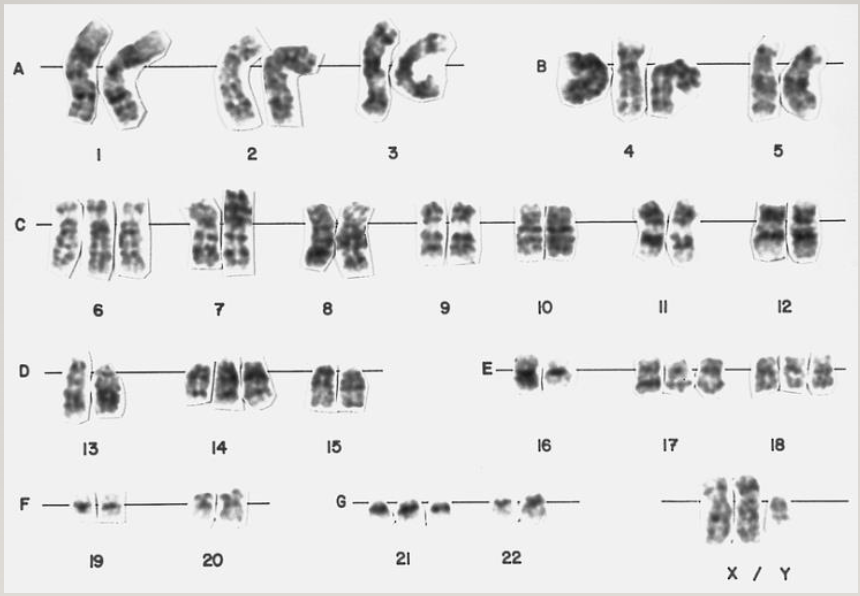
Karyotype: picture of chromosomes in an individual
Take cells, grow them, and treat them with chemicals that cause them to arrest at the condensation stage
Isolate the chromosomes and try to match them up
Each chromatid is a single DNA molecule
Two identical chromatids make a chromosome
Chromosomes in the cell cycle
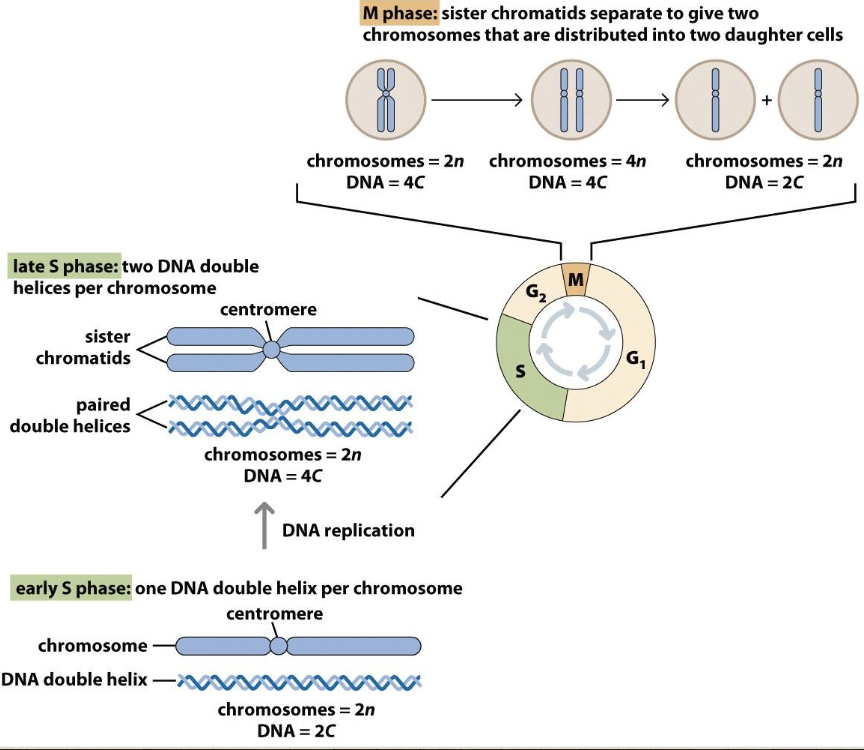
Checkpoints ensure chromosomes are made
In beginning of S phase, chromosomes have centromeres but aren’t paired
In late S phase, there are two DNA double helices per chromosome
In M phase, sister chromatids separate to give two chromosomes that are distributed into two daughter cells — you need 2 sister chromatids to divide
Chromosomes are critical to the accurate transmission and regulation of genetic information
Chromosomes are fundamental to the generation of variation
Incorrect meiosis, mitosis, or mutation can result in abnormal genetic composition
Stained chromosomes can be visualized and studied for major changes in chromosome structure
Chromosome structure
Types of chromosomes
The first chromosome has 2 sister chromatids, the second one only has 1 chromatid
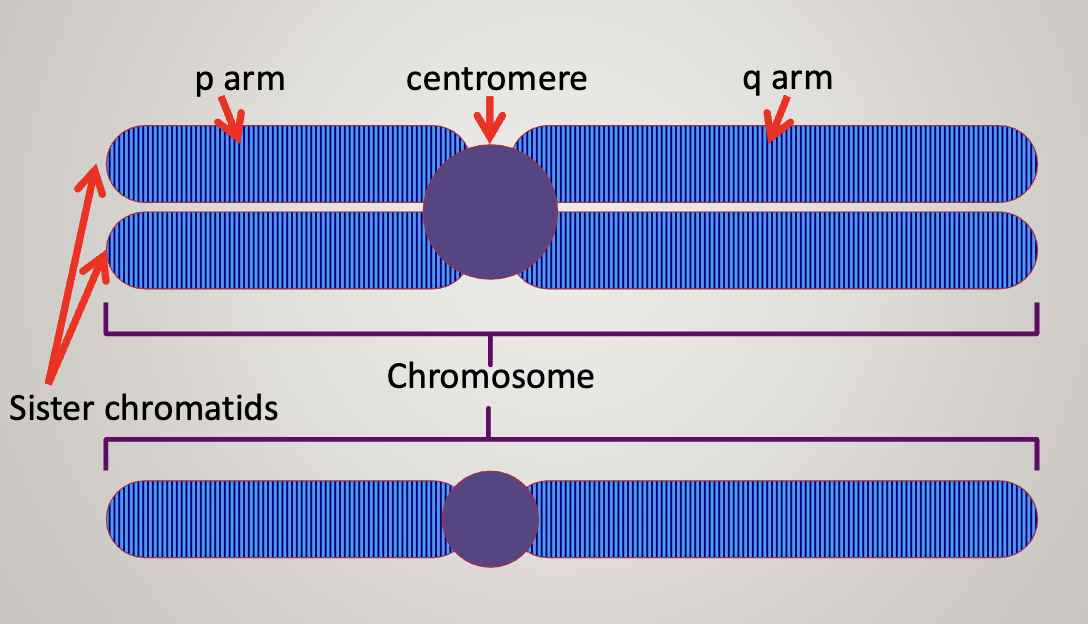
Centromere can be used as a landmark to describe the location of genes
P arm (short) and Q arm (long)
Chromosome is never in the exact center
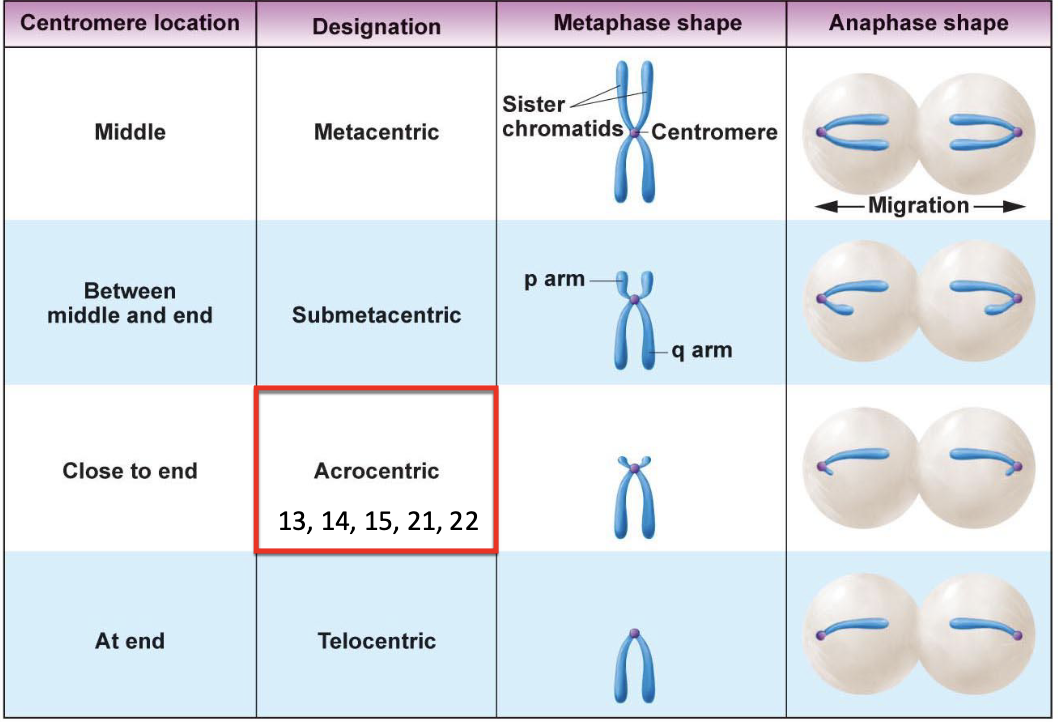
As the centromere goes to the end of the chromosome, the number of genes in the p arm goes down
Acrocentric chromosomes tend to have special functions (13, 14, 15, 21, 22)
Homologous chromosomes: one came from mom, one came from dad.
NOT IDENTICAL
Mitosis
Homologs do not pair
Sister chromatids separate during anaphase
The daughter cells are diploid (2n) like the parent cell
Meiosis
Meiosis I: Homologs pair and separate — recombination happens here
Meiosis II: Sister chromatids separate
Reduction division: the parent cell is diploid (2n), the daughter cells are haploid (n)
KNOW MEIOSIS AND MITOSIS
Meiosis I
Metaphase I: Homologs pair (p-p, q-q all along the chromosome)
Homologous chromosomes are driven to pair up
Crossing over also happens here (non-sister chromatids exchange genes from one homolog to another). It happens on average once every meiosis for each arm for each homologous pair
Recombination happens in a sequence-specific manner
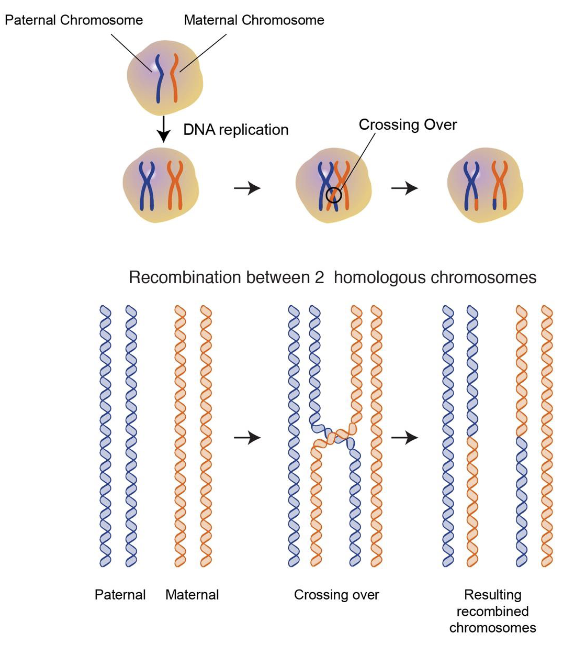
Anaphase: recombined chromosomes get pulled apart
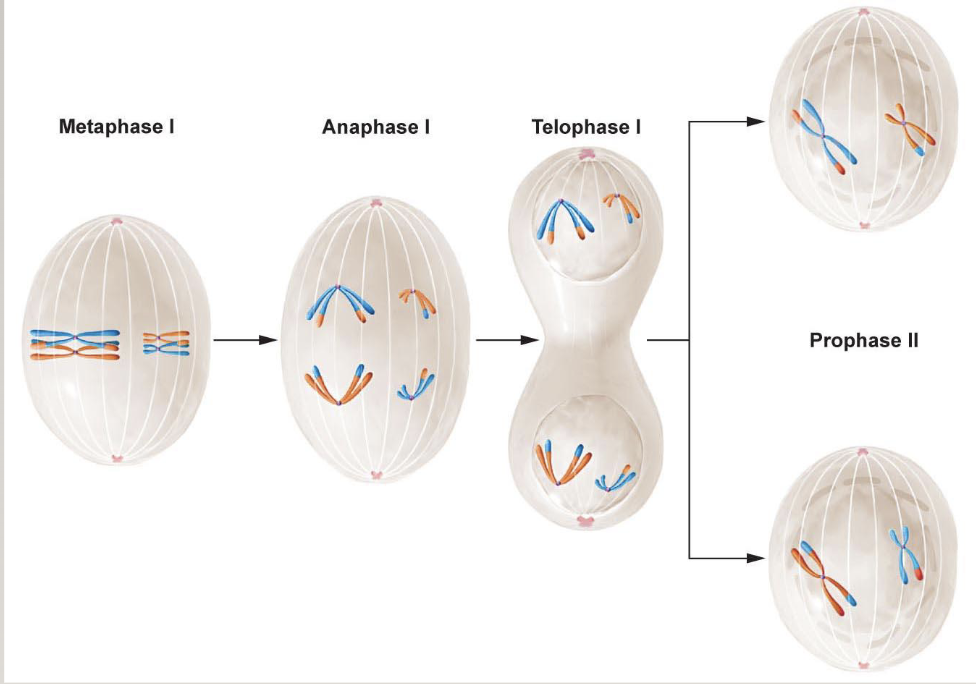
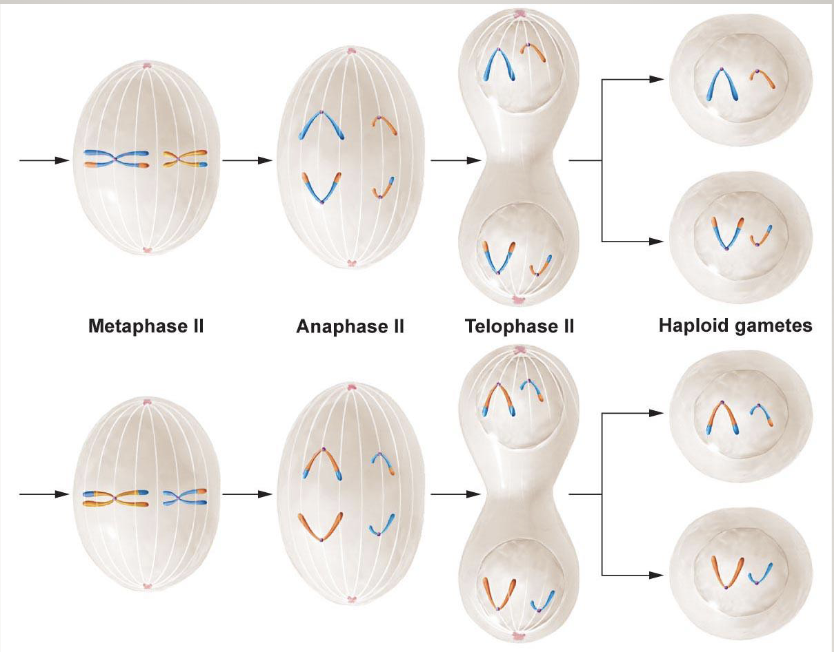
Meiosis II
Chromatids are separated out so gametes have only 1 set of chromosomes
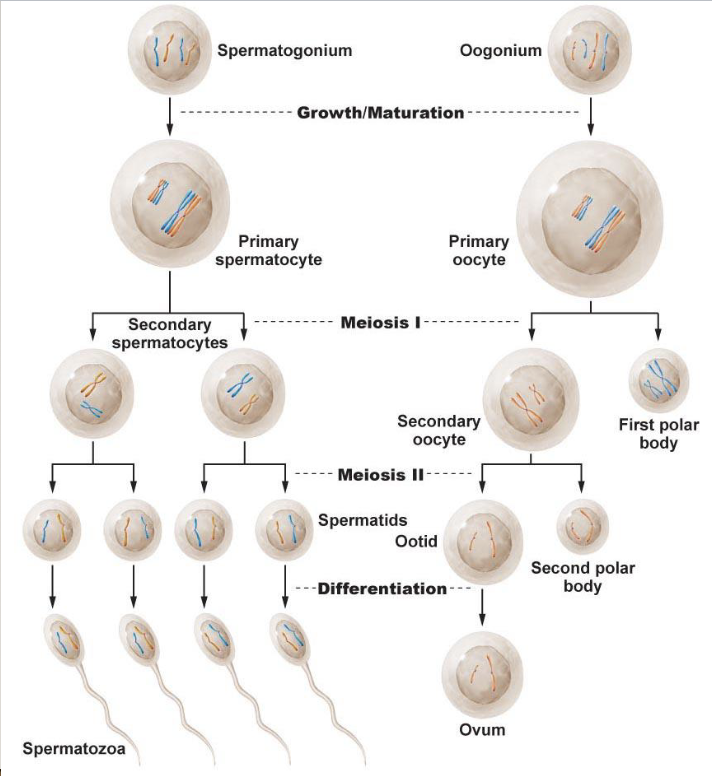
Spermatogenesis and oogenesis
Independent assortment
Genetic variation can be generated via independent assortment
Happens when homologs line up on the metaphase plate
A chromosome does not influence B chromosome
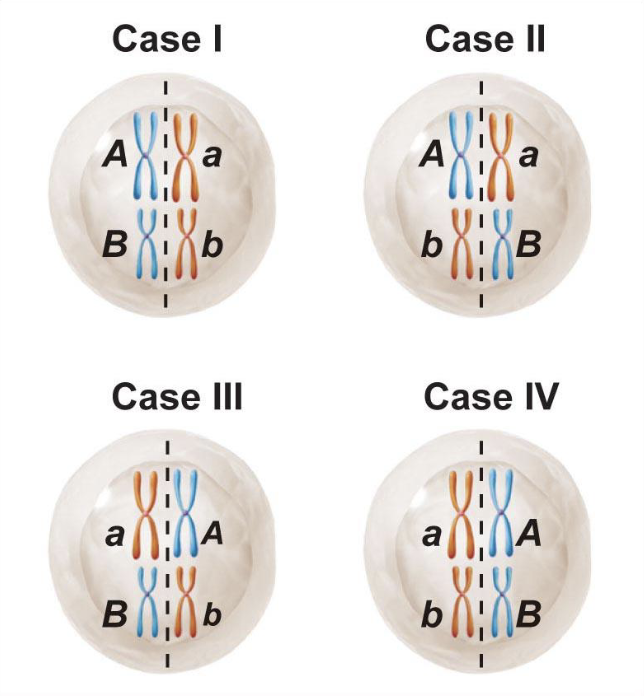
How are there so many combinations?

2²³ different sperm, regardless of recombination
Recombination
Mutation
Incorrect meiosis
Numerical chromosome abnormalities
Polyploidy: Extra whole sets of chromosomes (dispermy of fertilization of diploid cell)
Aneuploidy: one or more chromosomes extra or missing (non-disjunction)
Disomy: two copies of a chromosome, the normal state for most human cells
Trisomy: 3 copies of a chromosome
Monosomy: one copy of a chromosome
Mosaicism: when two cell lines with different karyotypes exist in a single organism
Mixoploidy: Two or more cell lineages mixed (mosaicism or chimerism)
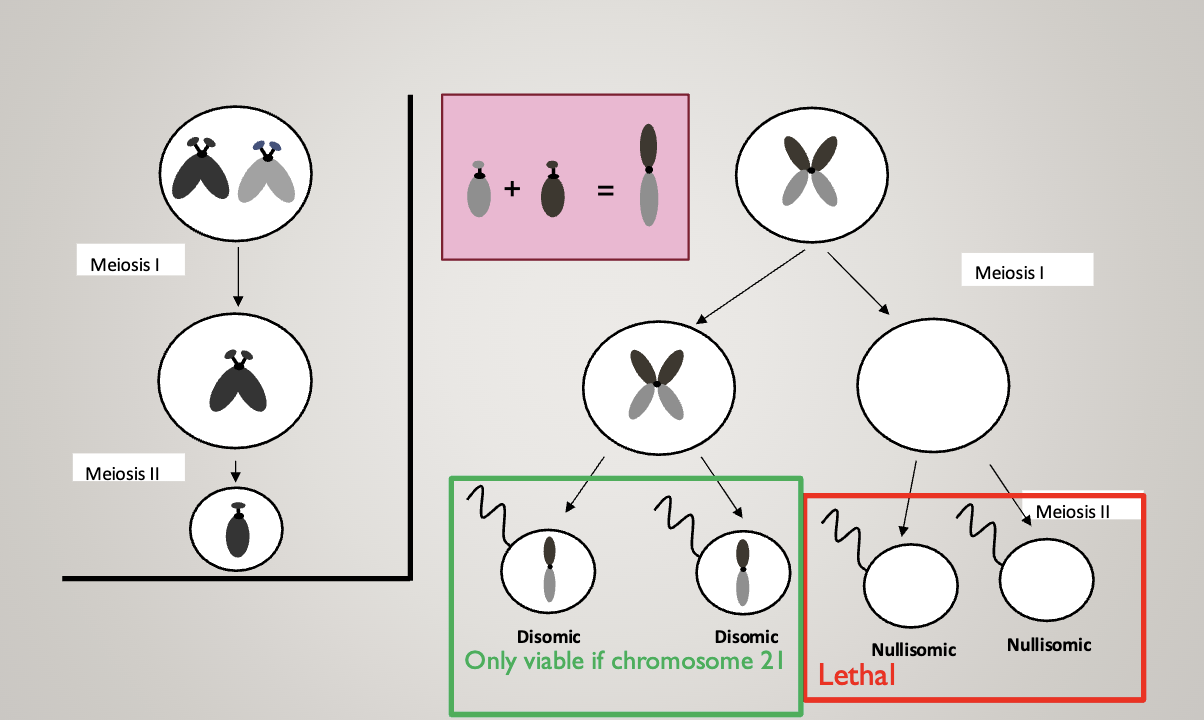
Non-disjunction:
Most aneuploidies result from meiosis I errors
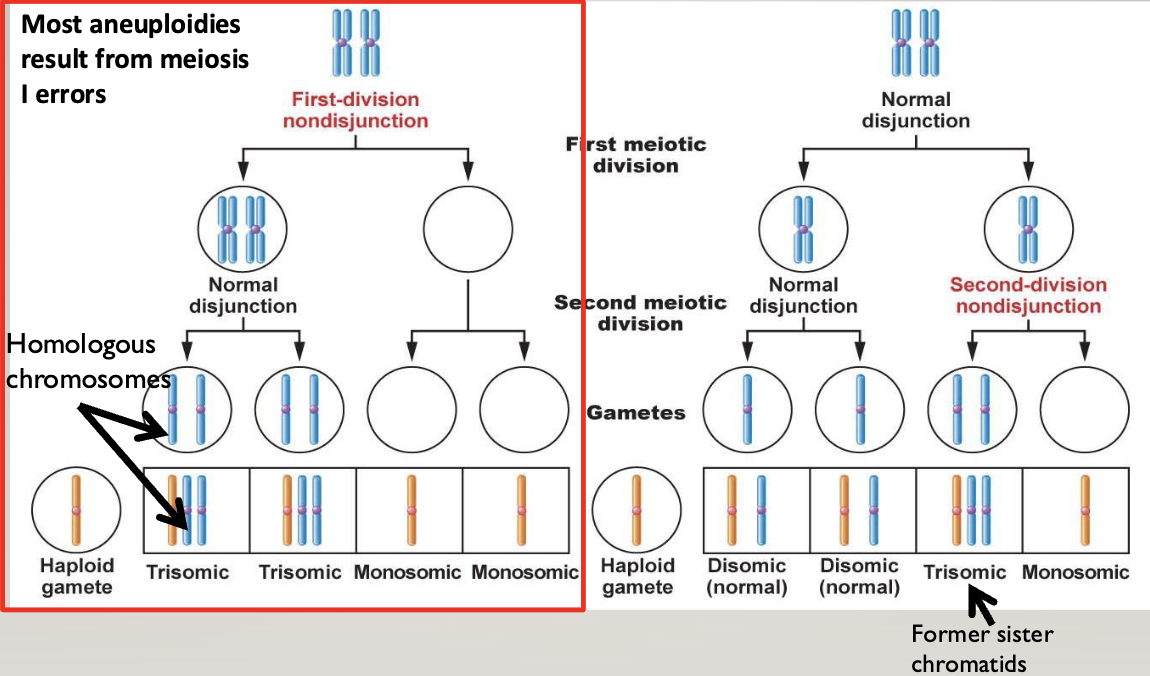
You end up with a cell with no chromosome, and a cell with both homologous chromosomes
In the second division, two gametes end up with 2 chromosomes (one mom and one dad), and two end up with nothing
In meiosis II errors, two gametes end up normal, one ends up with 2 identical chromosomes (both from mom or dad), and one has nothing
Chromosome mutations
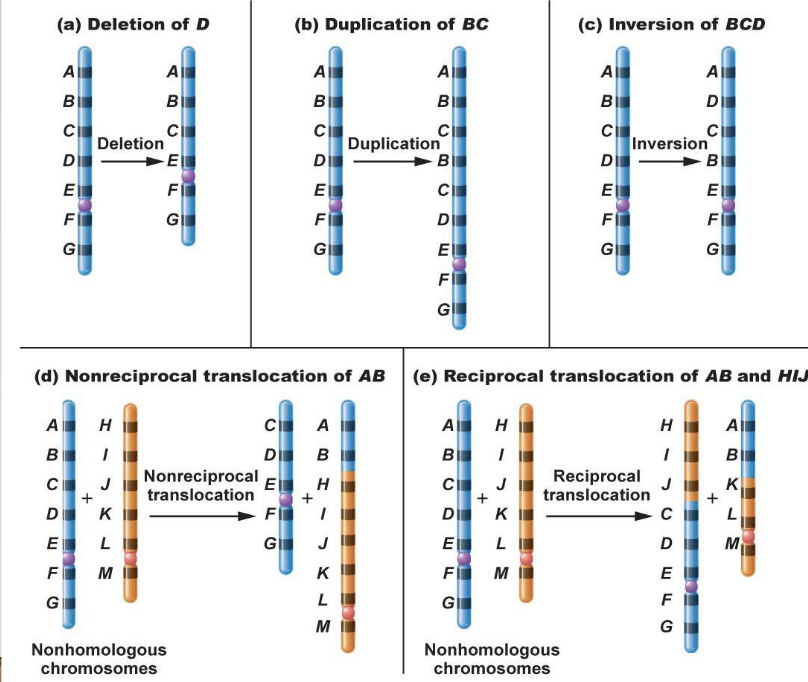
There are commonly humans with translocations
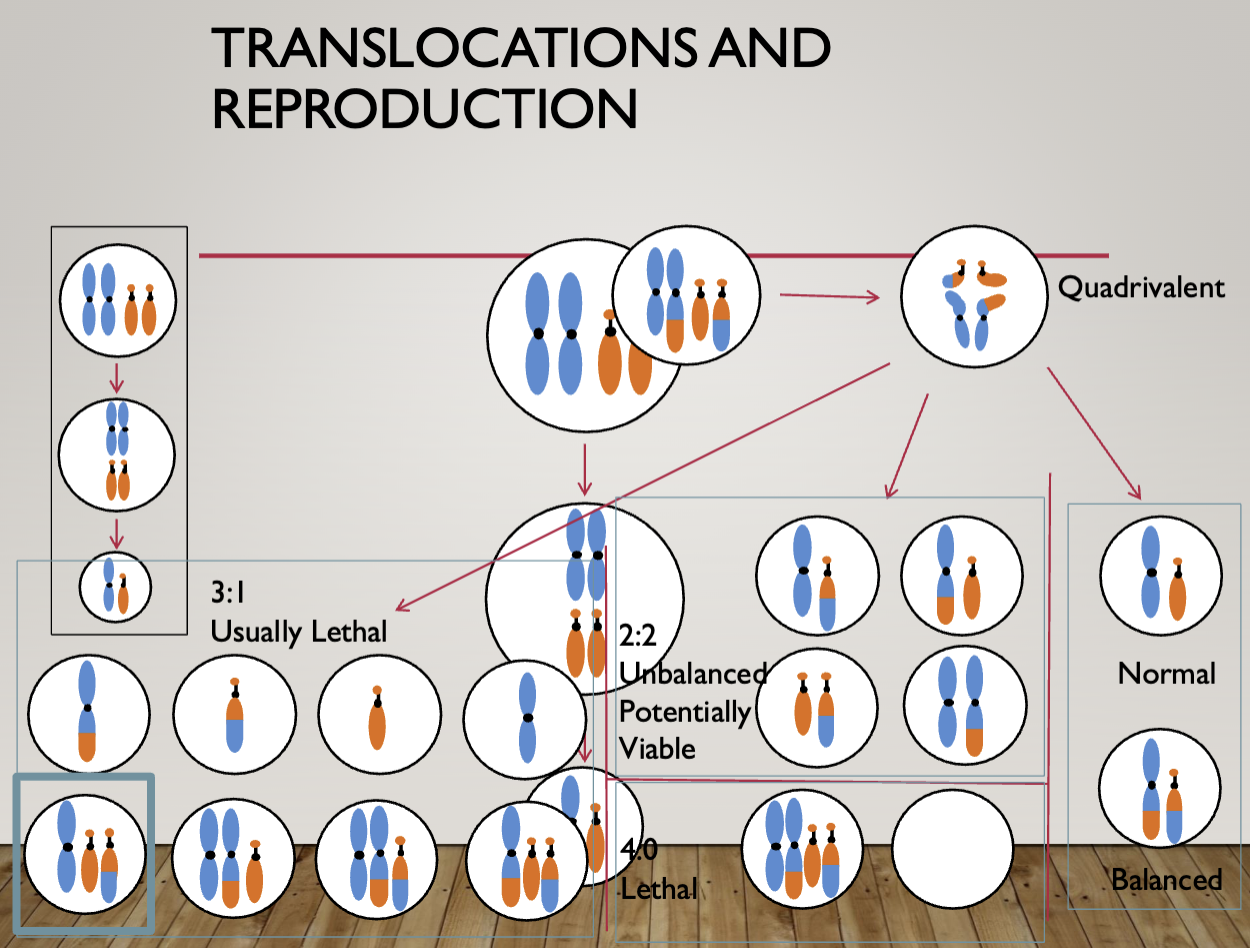
In reciprocal translocation, nothing is lost or gained as long as the breakpoint doesn’t disrupt the function
Produces 2 complete chromosomes, they just don’t look how they are meant to
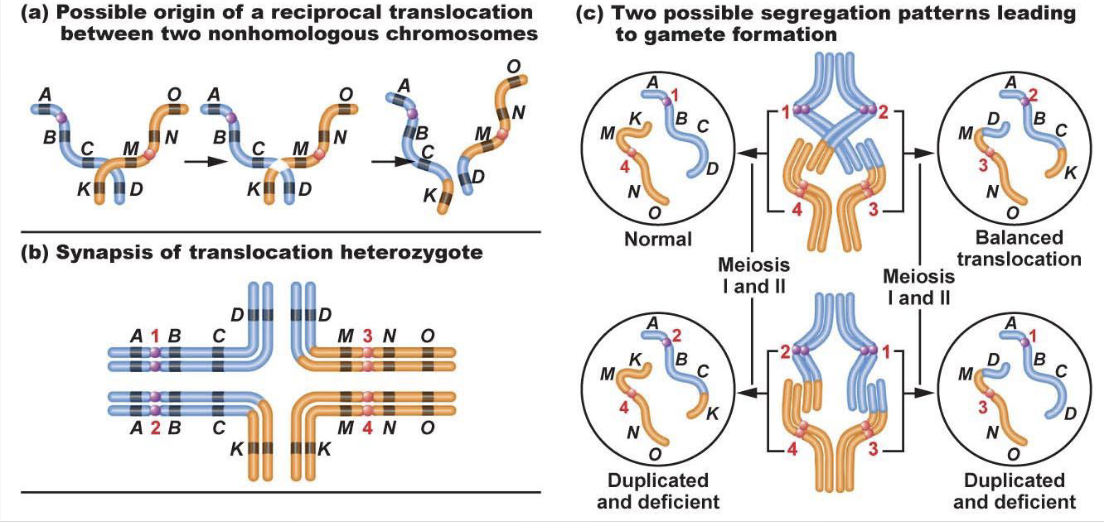
The drive for homologous chromosomes to pair is sequence-based
When there is a translocation, pairing up gets weird
Normal and balanced translocation will lead to a normal zygote
Leads to translocation carrier
Duplicated and deficient = some areas are duplicated while others are missing
1/23/2025
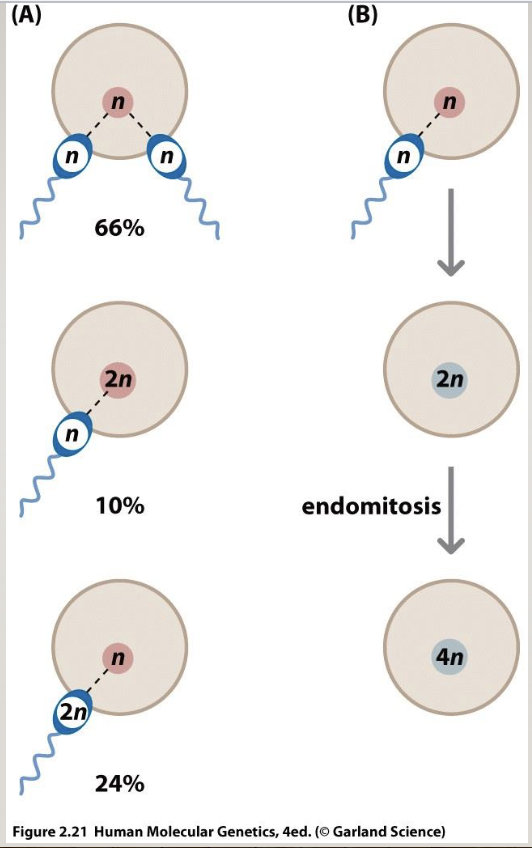
Origins of polyploidy
fertilization by 2 sperms
doubling chromosomes but not dividing (endomitosis)
euploid=normal
Fluorescence in situ hybridization (FISH)
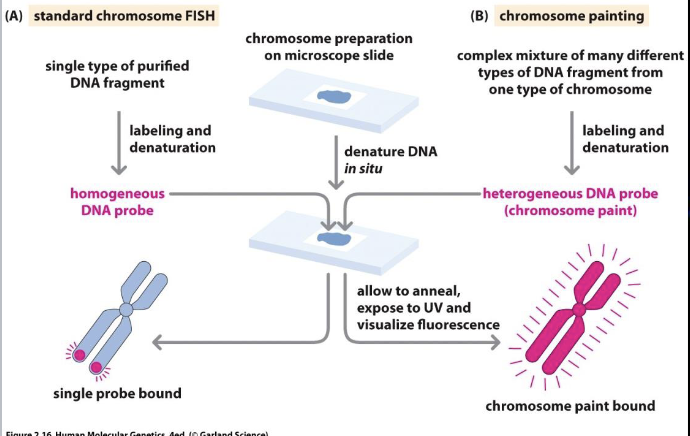
Good for quick chromosome count, detection of known/suspected deletions/deletions
Quick turnaround (24-48 hrs)
Limited bu probe availability
Targeted
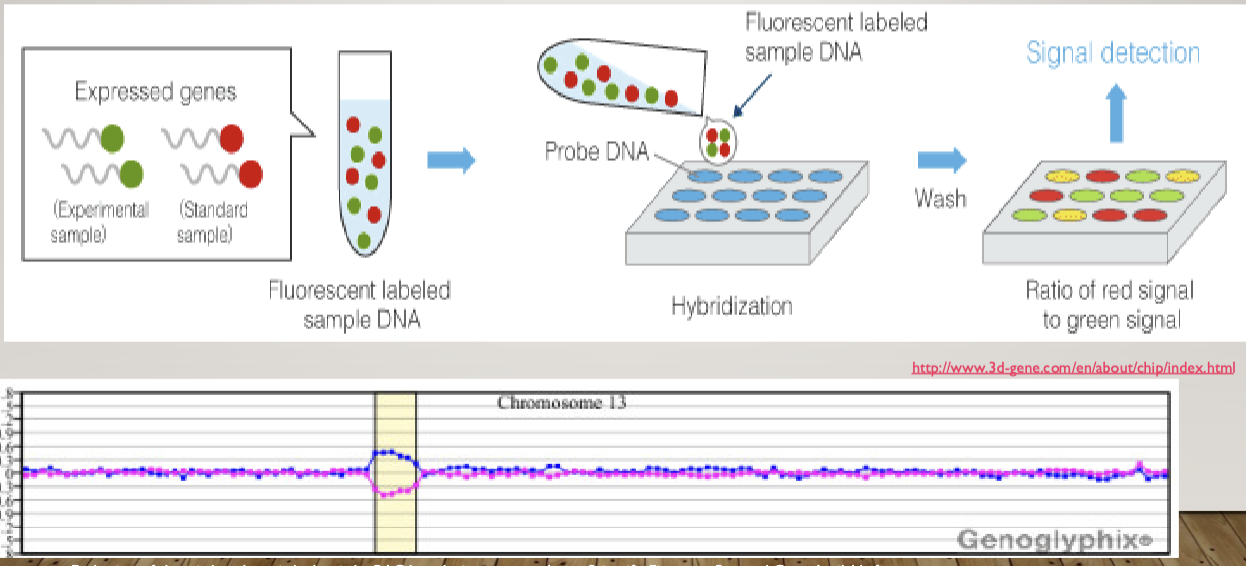
Microarray analysis
A way to measure the quantity of a given sequence of DNA in a sample
Can be used to detect full or partial aneuploidy including mosaicism
Standard testing during initial work up for a person suspected to have a genetic syndromeX$
Doesn’t work on translocation
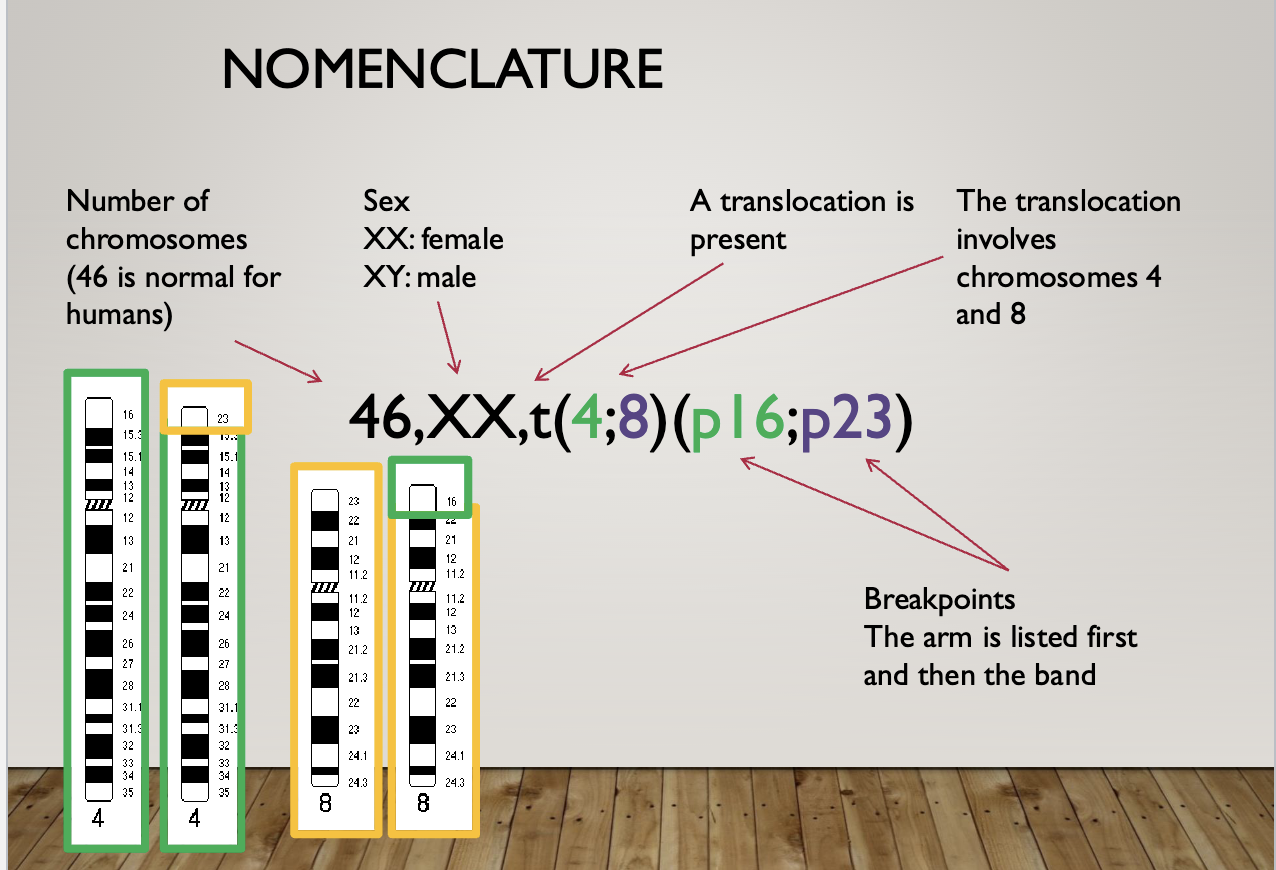
Nomenclature
46, XY = 46 chromosomes, male (normal)
47, XY+21 = 47 chromosomes, male, trisomy 21 (downs syndrome)
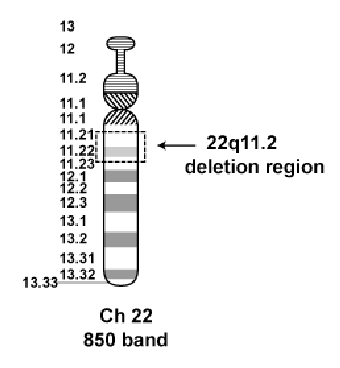
46, XX, del(22)(q11.2) = 46 chromosomes, female, deletion on chromosome 22 at band q11.2 (digeorge syndrome)
Common aneuploidies
Trisomy 21 (Downs syndrome)
Trisomy 18 (Edward syndrome)
Characterized by small size, unusual ear shape, small jaw, heart defects, usually lethal
Partial or mosaic form may be milder
Trisomy 13 (Patau syndrome)
Monosomy X (turner syndrome)
XXY syndrome (Klinefelter syndrome)
Trisomy 16
Triploidy
An extra copy of every chromosome
Most common polyploidy in humans
Lethal in non-mosaic state
Most commonly caused by diandry
Case studies
You are asked to evaluate Lucy, a 14 month-old girl with a history of
microcephaly, dysmorphic facial features, and developmental delays. In reviewing her family history you learn that she has two siblings both of whom have intellectual disabilities
You order a microarray for Lucy and get the following result...
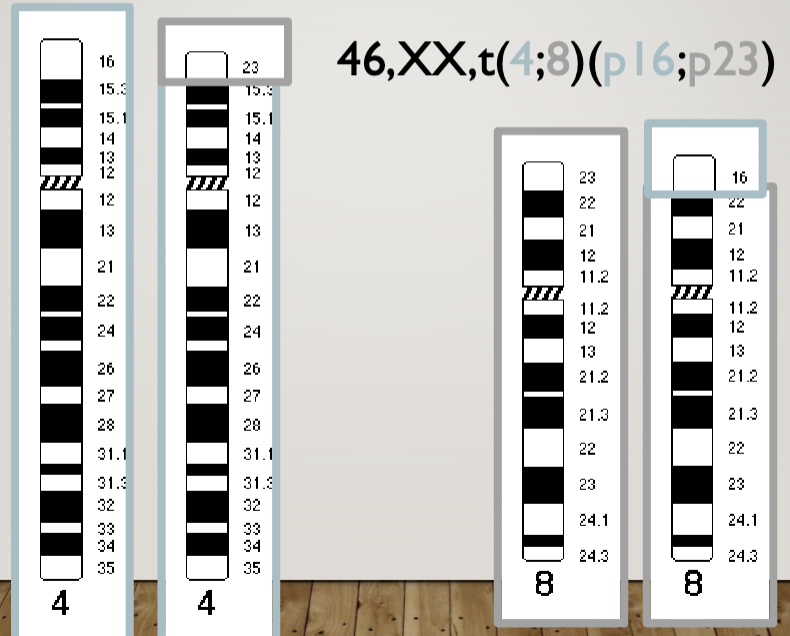
She has a 9.4 Mb gain on the p arm of chromosome 4 (partial trisomy) and a 6.8 Mb loss on the p arm of chromosome 8 (partial monosomy)
Seems like one of her parents had a translocation — she had a duplicated and deficient zygote
You then order a karyotype of the parents
Dad is 46, XY
Mother is 46, XX, t(4;8)(p16;p23)
Translocation in 4 and 8 at breakpoints 16 and 23 respectively
But she’s normal because she has all of her genes (translocation heterozygote)
Transfer of chromosomal regions between non-homologous chromosomes has an incidence of 1/500
The couple wants to have another child. What are their options?
Reproductive options
In Vitro fertilization with preimplantation genetic diagnosis
Gamete donation
Adoption
No kids
Post conception options
ultrasound
Chorionic villus sampling
Amniocentesis
Cell-free fetal DNA analysis
Translocations
Balanced: No gain or loss of genetic material at the translocation breakpoints
Unbalanced: Some gain or loss of genetic at the translocation breakpoints;
this may result in phenotypic anomalies
Translocation Carrier: an individual who carries a balanced chromosome
translocation; phenotypically normal (unless the breakpoints disrupt gene
function)
Origin of Translocations
Inherited: translocation carriers may pass the balanced (or unbalanced) form of a translocation onto their offspring
De Novo: a balanced or unbalanced translocation may arise through recombination in non-sister chromatids during (meiosis) or shortly after conception (mitosis)
Autosomal Reciprocal Translocation:
The exchange of segments between two non-homologous (heterologous) autosomal chromosomes
Balanced carriers are generally unaffected
Miscarriage risk: 20-30% on average for female carriers (but this varies greatly by translocation
Sex Chromosome Translocation:
The exchange of segments between one sex chromosome and another chromosome
The second chromosome may be an autosome or a sex chromosome (rare)
Generally causes infertility
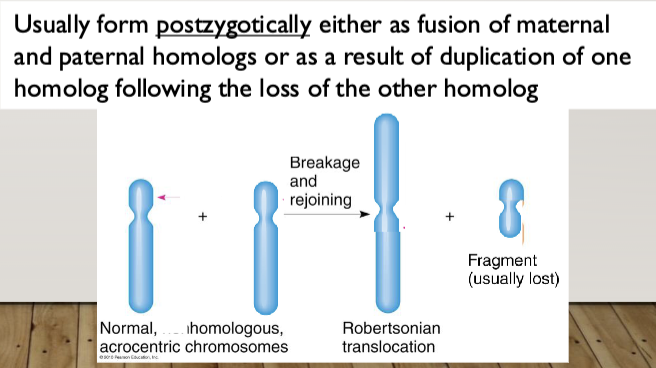
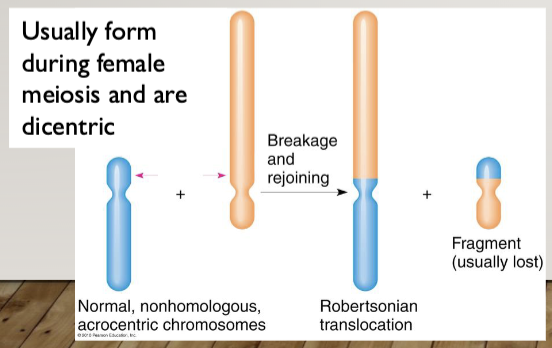
Robertsonian Translocation: the fusion of the long (q) arms of two acrocentric
chromosomes (13, 14, 15, 21, 22); the short arms (p) are lost
Heterologous Robertsonian: A Robertsonian caused by the fusion of the q arms of two different acrocentric chromosomes
Homologous Robertsonian: A Robertsonian caused by the fusion of the q arms of two copies of the same chromosome
Impact of translocations
Unbalanced translocations
Generally cause birth defects and intellectual disability
The larger the monosomic/trisomic segment the more severe the phenotype
Balanced translocation carriers
Generally healthy unless one of the breakpoints disrupts a gene
20-30% risk for miscarriage with each pregnancy but may be as high as 50+% (general
population risk is 15%)
The risk for miscarriage is generally higher if the translocation carrier is female
Males are at risk for oligospermia\
If a balanced de novo translocation is identified prenatally there is a 6% risk for an abnormal phenotype in the baby
IVF with PGD
IVF: collection of sperm and egg samples allowing for fertilization outside the uterus, promising embryos are implanted in the mother
PGT-A: testing done on embryos at the 8-cell stage, frequently via FISH, to look for imbalance, only balanced (normal or carrier) embryos are implanted
CVS and amniocentesis
Amniocentesis: 1/300 wish of miscariage, tests amniotic fluid
CVS: chronic villi, 1/100 risk of miscarriage
FISH may be used to obtain a preliminary result. Microarray is fast replacing karyotyping as the standard of care for prenatal samples
Cell-free fetal DNA analysis
Analysis of cell-free DNA in maternal circulation to assess for fetal aneuploidy
Case study 2
You are asked to see Pamela, a 27 year old pregnant woman who is currently
19 weeks. She had her fetal survey at her obstetrician’s office and the baby
was noted to have short long bones, an absent nasal bone, and pelviectasis.
She will be meeting with you prior to her targeted fetal anatomy ultrasound
to discuss the findings and her options. Her husband, Trevor, accompanies her
today
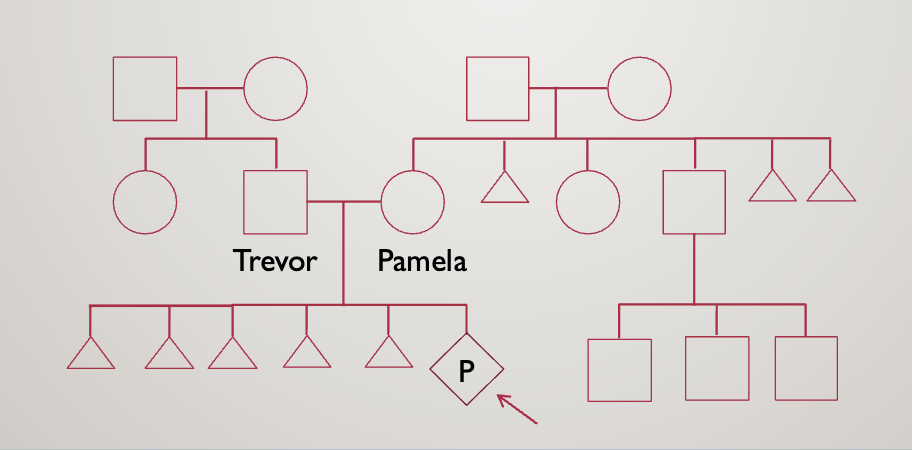
They opt for an amniocentesis
Result: 46, XY, rob(21;21),+21
Rob=robertsonian translocation (acrocentric, one chromosome is twice as long)
Leads to Down syndrome
They then opt for karyotyping
Pamala is 45,XX,rob(21;21)
This means she has just 1, really long 21st chromosome
She will always have nondisjunction — can only make gametes with no chromosome 21 and ones with 2
1/28/2035
Mendelian genetics
Key concepts
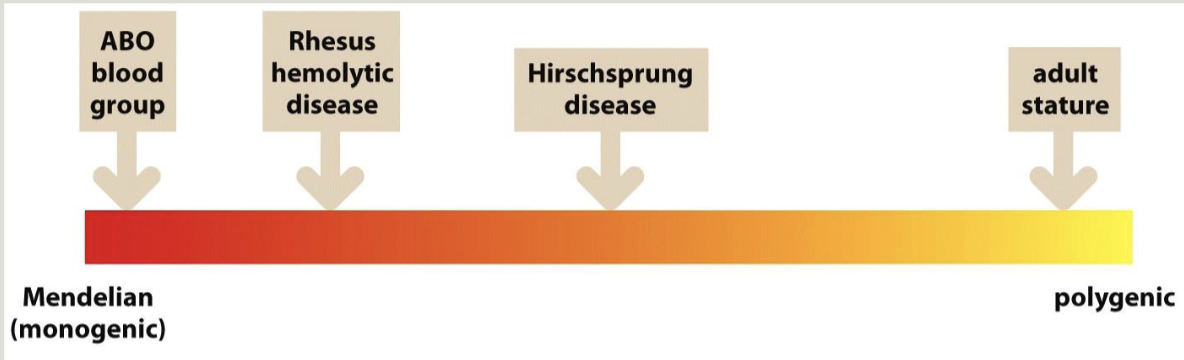
Inheritance of a characteristic follows mendel’s rules if its presence or absence is determined by the genotype at a single locus
Mendelian characteristics can be dominant, recessive, or co-dominant. A characteristic is dominant if evident in a heterozygote
Human Mendelian characters give pedigree patterns that are often recognizable
Most characteristics are actually polygenic (multifactorial)
Variable expressivity: a trait or disease can manifest differently among affected individuals
Phenocopy: a phenotype that mimics the trait of interest but does not share the genetic etiology
Disease alleles of genes are rare
Subject to certain conditions, allele frequencies and genotype frequencies are related by the Hardy-Weinberg formnalle=a
p+q=1
p²+2pq+q²=1
Inheritance patterns
Autosomal recessive
Usually only one generation affected
Carriers are not affected
No association with sex
Children of a carrier couple are at 25% risk
Unaffected children have a 2/3 chance of being a carrier
Parents of a recessive child are obligate carriers
Consanguinity increases the risk for AR conditions
Fancy word for incest
Examples

Autosomal dominant
Multiple successive generations (but may arise de novo)
NO association with sex
Children of an affected individual at 50% risk
Examples
Huntington’s disease
Treacher Collins syndrome
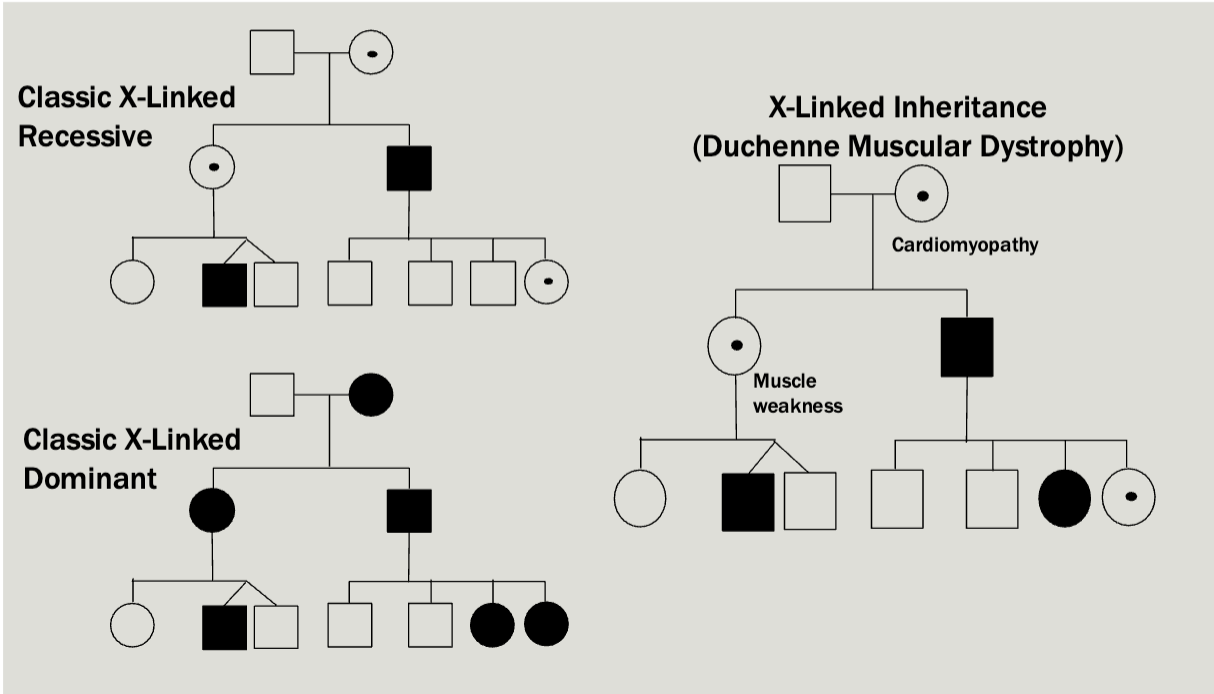
X-linked
Multiple generations affected
Males tend to be more severely affected than females
Can be dominant or recessive but usually there is a broad spectrum
All daughters of affected males will be caries/affected
No male-male transmission
Each child of an affected female is at 50% risk
Examples

Pedigree is maybe a little unexpected — Mosaicism makes it so that women
will sometimes express the diseased X
Which X (the paternal or maternal copy) is chosen for inactivation in an XX embryo is determined randomly and independently by each cell of the embryo, at the 10-2 cell stage
However, once a cell has chosen which X to inactivate, that X remains inactive in all its daughter cells
Barr bodies
XY males keep their single X active (no Barr body)
XX females inactivate one X in each cell (one barr body)
Females with turner syndrome (45,X) do not inactivate their X (no Barr body)
Males with Klinefelter syndrome (47,XXY) inactivate one X (one Barr body)
47,XXX females inactivate two X chromosomes (two Barr bodies)
Y-linked
Only present in Males
Only male-male transmission (females cannot be carriers)
All sons of affected male will be affected
Super rare
Example
Spermatogenic failure
Mitochondrial
All affected children of an affected female will be affected (regardless of sex)
Affected fathers will not have affected children
Significant variation in severity with a tendency to get worse with each generation
Mitochondrial diseases caused by mutations in the nuclear genome follow an autosomal pattern of inheritance
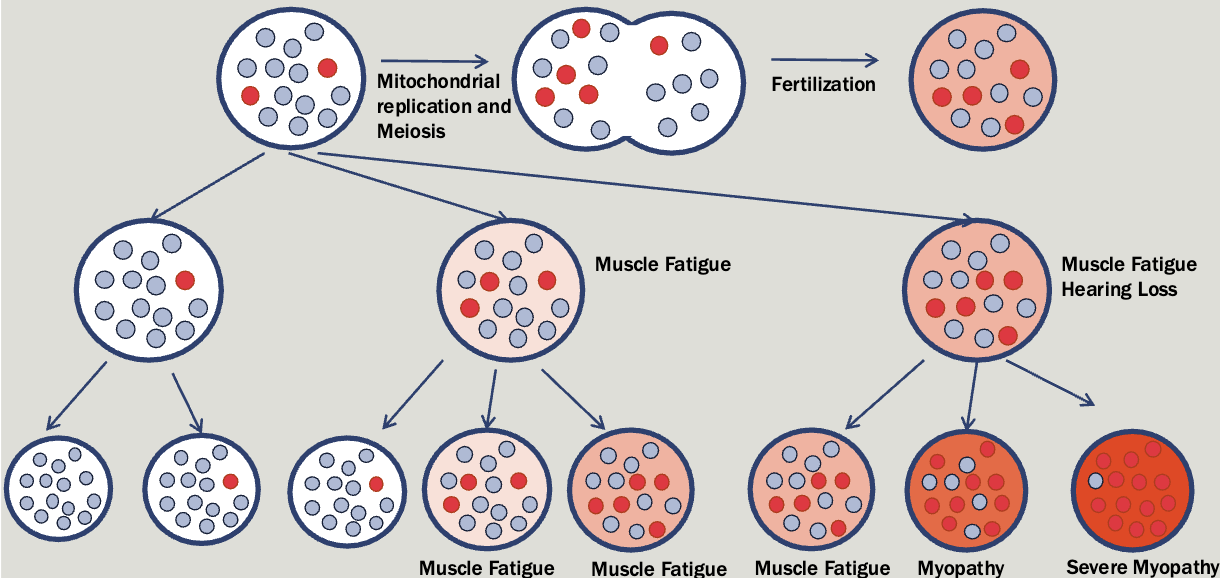
Mitochondrial genes
MELAS
Leigh syndrome (also in nuclear genome)
Example: mitochondrial myopathy
Treatment
Mitochondrial replacement
Mendelian characteristics vs genes
Locus heterogeneity: Sam clinical phenotype from mutations at any one of several different loci. Also called clinical heterogeneity
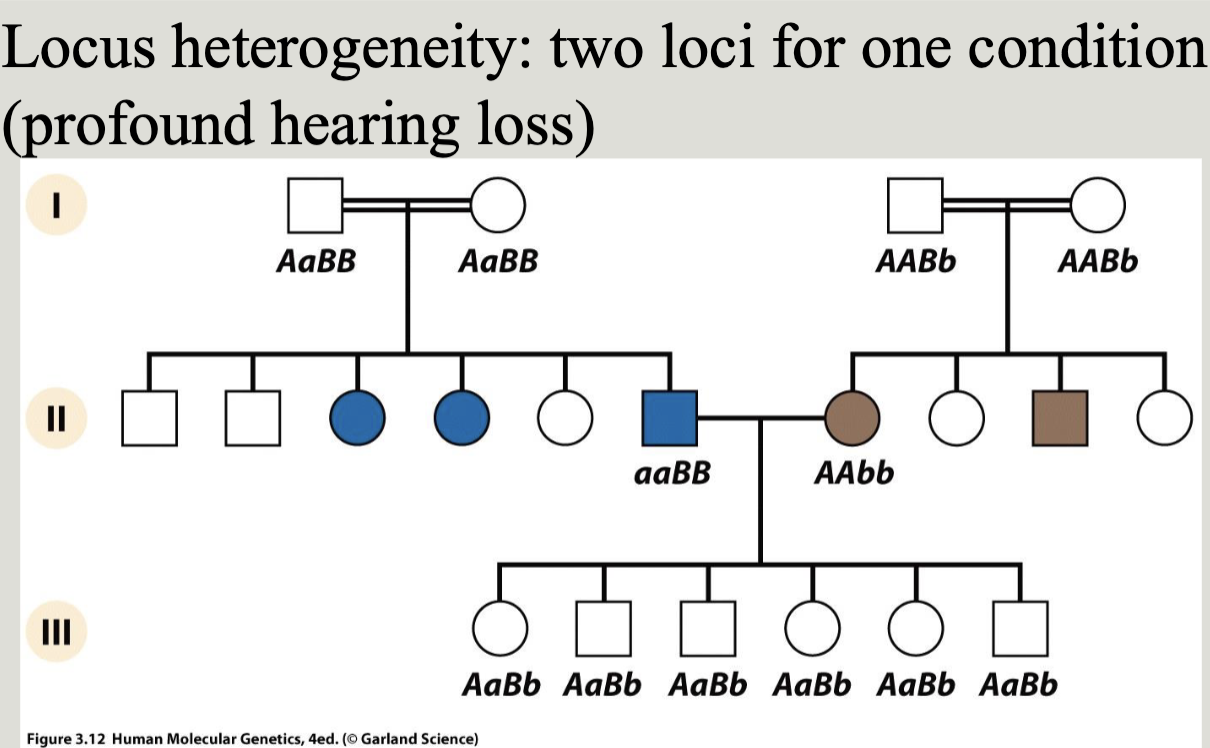
Allelic heterogeneity: different mutations at the same locus result in the same clinical phenotype i.e. different mutations in the same gene may cause loss or gain of function leading to a specific mendelian disease
Mendelian complications
Non-paternity
When the stated after is not the biological father
May be uncertain
Difficult to address with families
Ascertainment bias
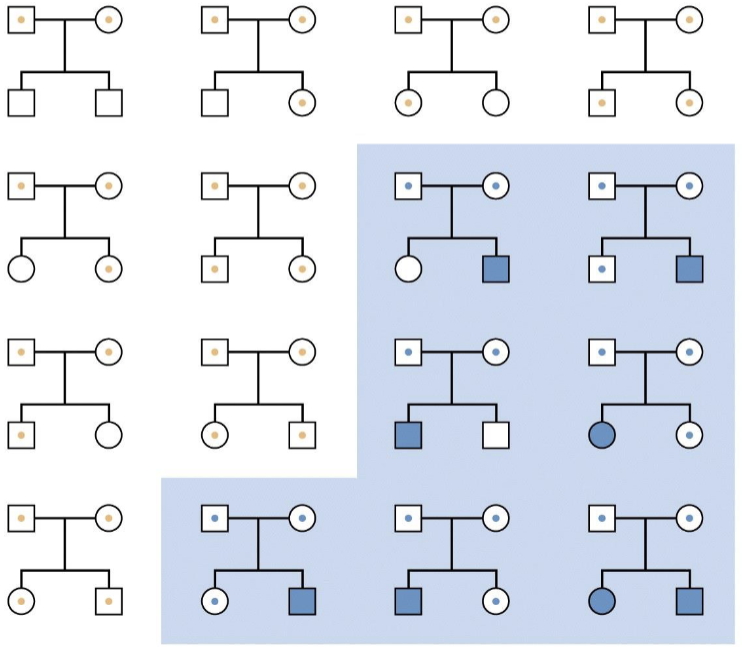
Not ¼ but 8/14
Common recessive looks like a dominant
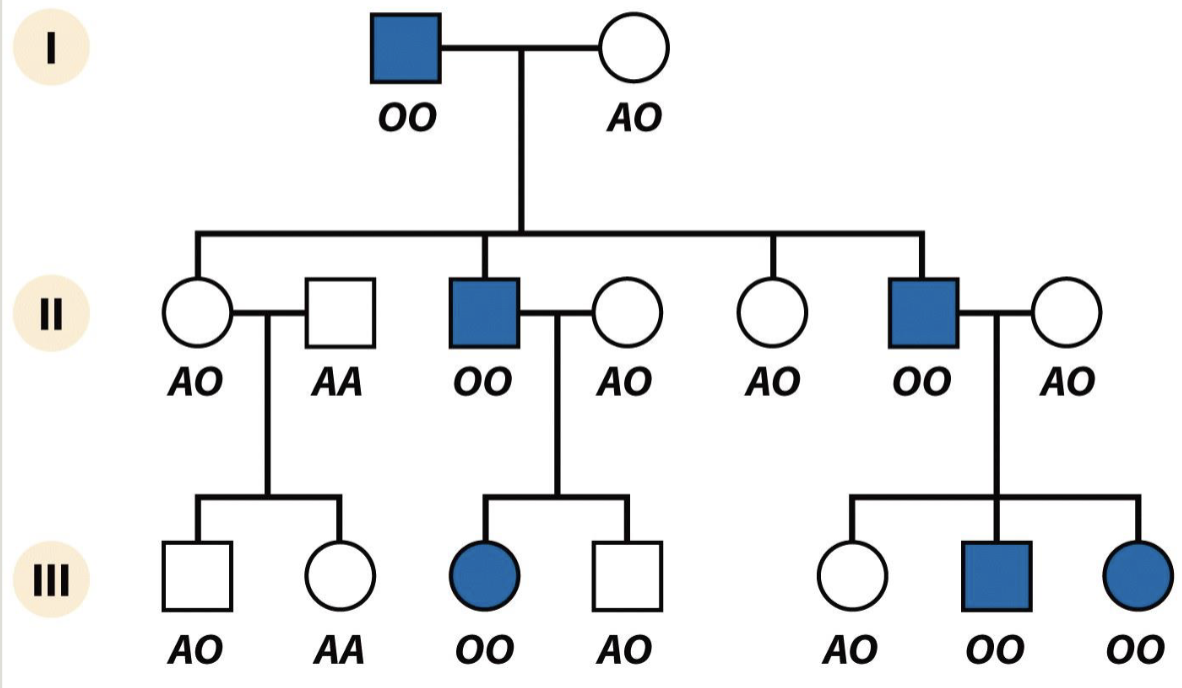
O is recessive but also really common
Non-penetrance
Parent has condition but it doesn’t show up in the phenotype
New mutations and mosaicism
Key concepts
Gonadal mosaicism
When the gamete population of an individual is mosaic for an allele
Can cause an unaffected individual to have multiple children with an autosomal dominant condition
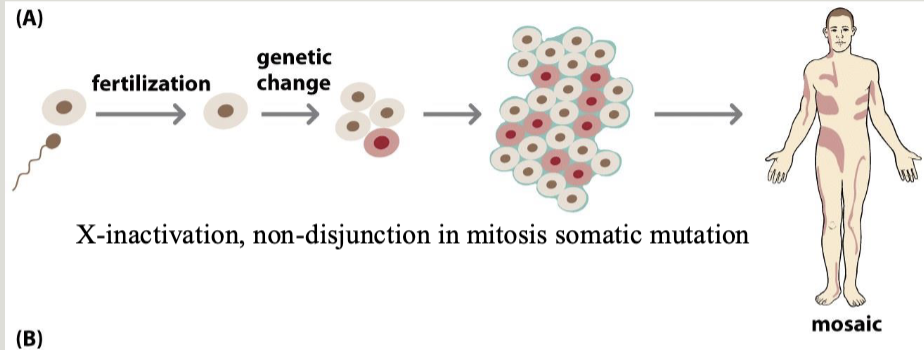
Chimerism
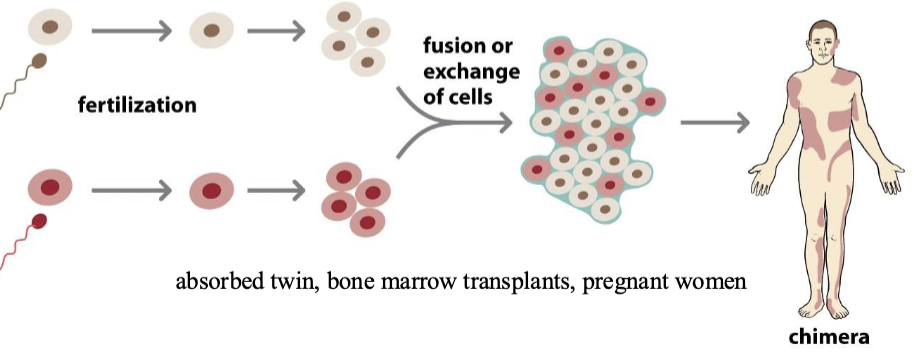
1/30/2025
Case study 1
You are as ked to evaluate Alex, a 2 year old boy with a history of easy bleeding and bruising. As the family is very wealthy and influential you agree to make a house call. At the family home you meet Alex’s parents, Lexi and Nick, and his four older sisters. Lexi repor ts the following family history
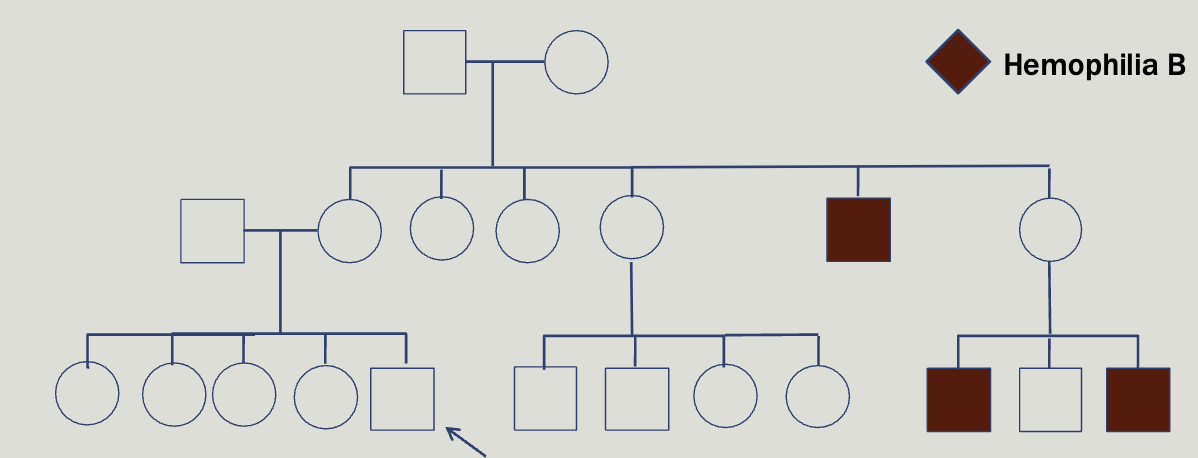
Hemophelia is x-linked, however about half of cases result from de novo mutation
Deficiency of clotting factors 8 and 9, found at F8 and F09 genes
Locus heterogeneity
You can test factor 8 and 9
Not able to diagnose 90% of female carriers
Female carriers can be symptomatic and may be at-risk for bleeding especially during childbirth
Case study 2
Maryann, a 45 year old woman, comes to yo ur clinic to request testing for a familial condition that causes dementia and involuntary movements . She reports that her father w as diagnosed at age 63. Her brother was also diagnosed 5 years ago
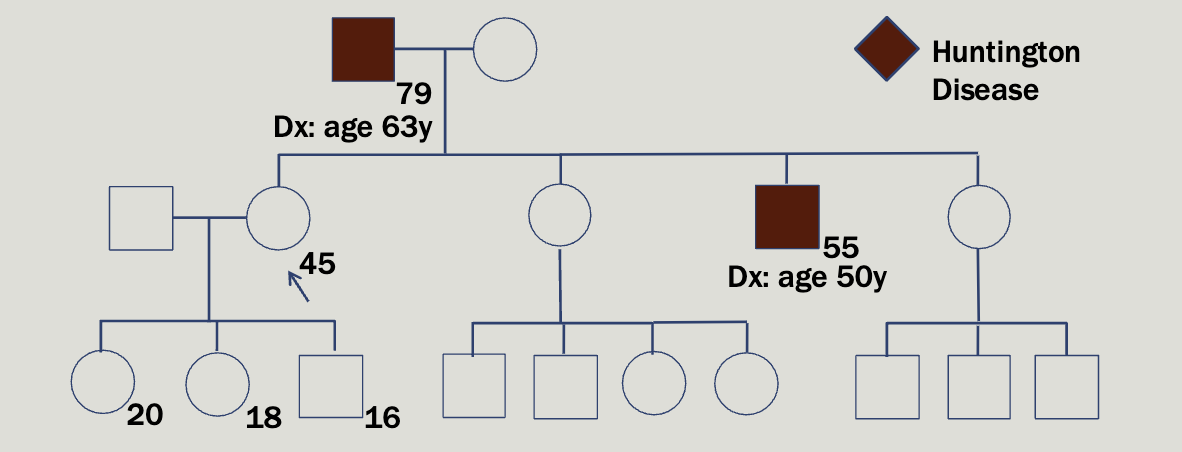
Huntington is autosomal dominant and found on chromosome 4
Caused by a CAG repeat expansion in the HTT gene
3 nucleotides are repeated over and over again — highly unstable
Normal alleles: 26 or fewer CAG repeats
Intermediate alleles: 27 – 35 CAG repeats; will not develop HD but
may have children with HD due to anticipation
Reduced-penetrance alleles: 36 – 39 CAG repeats; some individuals
in this range will never develop symptoms
Full-penetrance alleles: 40+ CAG repeats; 60+ CAG repeats may
result in juvenile HD
Increasing repeat size is associated with earlier onset of symptoms
Easy to test for
How did we figure out where the huntingons’s allele was?
Crossing over
Need a lot of families where the disease incidence is high
In one part of venezuela, there was a small founder population, and now the incidence rate is 1/125
Case study 3
Omar and Nadia come to your clinic to discuss a family history of intellectual disability and dysmorphic facial features. Nadia is 28 weeks pregnant
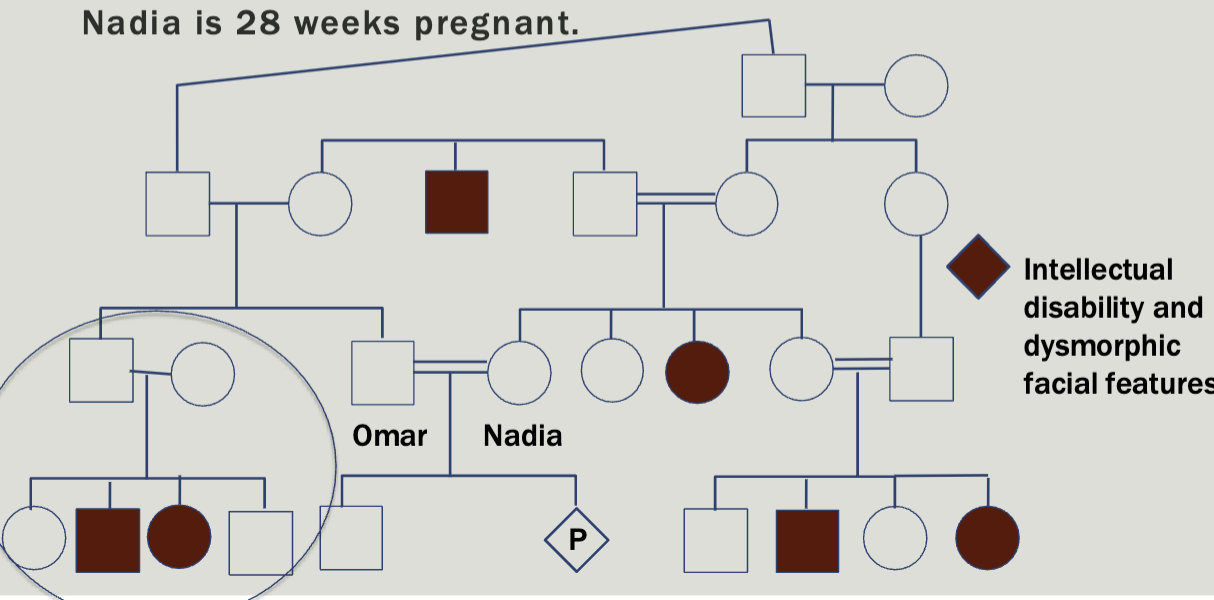
Autosomal recessive
Consanguinity increases the chance for homozygosity by descent
Everyone has about 5-7 deleterious recessive alleles
Consanguinity increases the chance to have a child with an autosomal recessive condition
Increases the risk for birth defects that are not part of a
known genetic syndrome
Case study 4
Sally’s daughter, Claire, has been diag nosed with a rare, autosomal recessive condition called Random syndrome. Sally and Claire’s father are divorced. Sally and her new husband, Bernard, have just learned they are pregnant and are worried about the risk that their baby will also have Random syndrome.
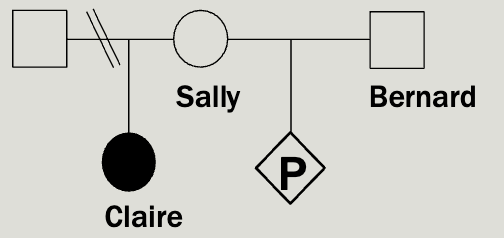
You know that that incidence of random syndrome is 1/14,400 but a
carrier frequency has not been reported.
Provide Sally and Bernard with a recurrence risk estimate for their baby.
To calculate the risk to a pregnancy for an autosomal recessive disease you multiply
the chance that each parent is a carrier by the ¼ chance that two carrier parents would both pass on the mutation (which is always ¼).
We know that Sally has to be a carrier
The incidence rate in the population is 1/14,400
1×bernards chance of being a carrier×1/4
Practical hardy-weinberg
The carrier frequency of an autosomal recessive disease in the general population can be determined from the disease incidence

So Bernards chance to be a carrier =2pq

Since p+q=1, and q is so small, we can assume that p=1

Probability that the baby is a carrier = 1×1/60×1/4
1 in 240
Ethnicity-based carrier screening
Northern European Caucasian — cystic fibrosis
French Canadian and Cajun — Tay Sachs disease
Ashkenazi Jewish ancestry
Much of the US Jewish population
Evidence suggests that the Ashkenazi Jewish population bottlenecked and experienced founders effect
Dor Yeshorim
International genetic screening system
Adults submit a blood sample tested for common autosomal recessive diseases
each participant receives an identification number but does not get the results
When two people are considering an engagement or when a rabbi is attempting to arrange a marriage, the identification numbers can be submitted to determine if the couple is compatible (not carriers for the same condition)
Pan-ethnic carrier screening
Screening for many different genetic conditions regardless of ethnic background
2/4/2025
Birth Defects
Birth defect = congenital anomalies
It can be major or minor
3-5% incidence rate
Causees
Sporadic: usually isolated within a family, unknown etiology, not expected to be inherited
Chromosomal syndromes
Genetic syndromes
Teratogens
Positional
Vocab
Isolated: not associated with other anomalies, etiology might be known, may be inherited
Syndrome: a well-known pattern of anomalies believed to have the same cause, often genetic/chromosomal but may be environmental
Sequence: a group of related anomalies believed to be caused by a single major anomaly that alters the development of other structures
Common major anomalies
Heart defects
1/200
Usually sporadic
Neural tube defects (spina bifida)
1/1000-1/200
Usually sporadic
May be environmental
Club food
1/1000-1/500
May be positional
Pyloric stenosis
1/500 males, 1/1000 females
Clef lip
1/1000-1/500
Usually sporadic by also associated with many genetic and environmental syndromes
Gastroschisis
1/10000
Usually sporadic, may be related to maternal age or smoking
Common minor anomalies
2,3-toe syndactyly
Occular anomalies
Hypoterlorism/hypertelorism (eyes being close together or far apart
Syndrome vs sequence
Syndrome: Achondroplasia
Dwarfism
Comes with short limbs, frontal bossing, low nasal bridge
FGFR3 gene mutation
Sequence: Oligohydramnios sequence
Potters sequence
Limited lung development, joint conttractures
Low amniotic fluid causing fetal compression
Symptoms are all the result of one abnormality
Heirarchy!
Organelles
Peroxisomes
Participate in energy metabolism
Replicate independently of the cell but do not have their own genome
Break down uric acid and other waste products
Lipid biosynthesis
Cause Zellweger syndrome
Autosomal recessive
12 peroxin genes (locus, allelic, and clinical heterogeneity)
Low muscle tone, dysmorphic features, seizures, lover disease, usually die within the first year of life
Cellular structure
Demyelinating diseases
Loss of the myelin sheath
Neurologic deficits due to failure to transmit signals across the neuron
Can be reversible
Causes:
B12 deficiency
Tay Sachs disease
Biral infection
MS
Alcholol
Skin Cells
Epidermis, dermis, and hypodermis
Protects against environment
Sensory
Fat storage
Vitamin production
Epidermolusis bullosa
Extreme skin fragility with blistering
Healing around the digits may lead to fusion of the fingers and toes
Mucosal tissues may also be involved
Teeth and nails fall out
embryology
The zygote in each cell in very early stage vertebrate embryos can give rise to every type of adult cell
Stem cells
Fetal alcohol syndrome
Effect dependent upon exposure amount an timing
Changes to facial features
Intellectual disability
Case study
Melody and Kevin have just given birth to their second child, a
daughter named Fern. The family has recently moved
to the area and Melody’s prenatal records have not yet been
transferred. The couple shares with you that they were told that
their baby would have cleft lip and palate and might have other
concerns as well. You have been asked to evaluate the baby
MRI shows semi lobar holoprosencephaly
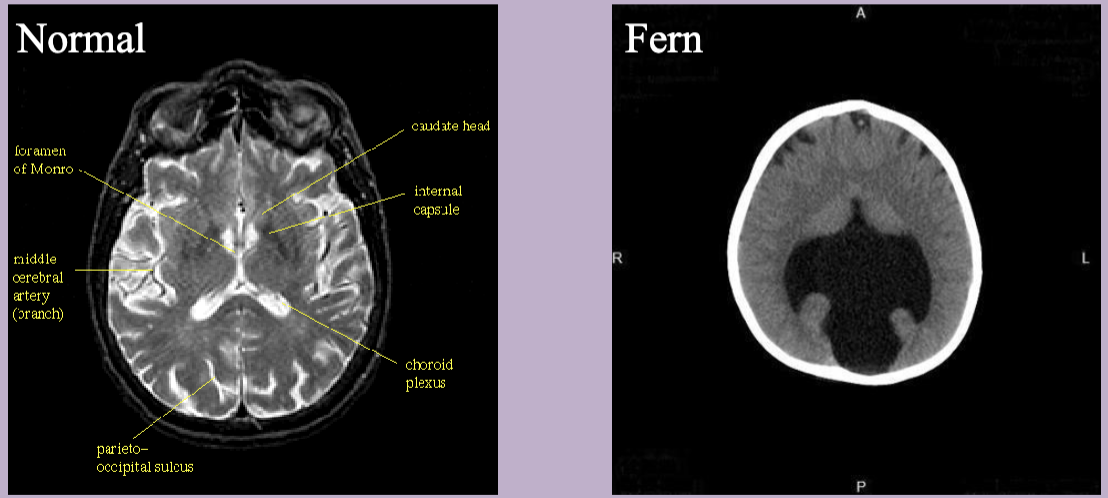
Holoprosencephaly
Abnormal development of the forebrain leading to incomplete separation of th cerebral hemispheres
2/5/2024
Genetic variation
Genetic variation is measured at 2 levels
Within a population (among individuals)
Consider population history/current size
Big populations tend to have more diversity
Populations that have gone through a bottleneck tend to have less diversity
Among populations (of the same species)
Consider gene flow/connectivity
Types of genetic variation
Chromosomal
Karyotypes describe what your chromosomes look like
Sex chromosomes vs autosomes
Haploid = n
Diploid = 2n
heterogametic = different sex chromosomes (XY)
homogametic = same sex chromosomes (XX)
Variation in chromosome number can be problematic for inter-breeding
horses: 2n=64, n=30
donkeys: 2n=62, n=31
mule: 2n=63, no n — infertile
one chromosome can’t pair up in meiosis
But things get weird! sometimes hybrids with odd numbers of chromosomes can reproduce
Chromosome variation is problematic during meiosis
Genetic information does not align during crossing over; interferes with gamete production
Chromosomes can fuse together, so you can have species within the same genus with very similar DNA sequences but different numbers of chromosomes
Polyploidy: Have different numbers of sets of chromosomes
Chromosomal rearrangements
Inversions
Chromosomal inversion separates annual and perennial ecotypes of monkey flower
Adaptive differentiation
Translocations
Mitochondrial DNA
Useful for phylogenetic questions
Only maternally inherited
Eggs are big, sperm are small
There are many more mitochondria in the egg
It doesn’t vary much between individuals of the same family
Can be useful for separating groups
Whole genome acts as one locus
You can use mitochondrial DNA as a marker when you can’t with nuclear DNA bc you will always have more copies of the mitochondrial DNA
Used to look at differences between Ridley Sea Turtles across the world
Can also be used to look at differences among species (phylogenetic perspective)
Chloroplast DNA
Useful for phylogenetic questions
Only maternally inherited
Chloroplast DNA variation resolved species relationships in the carrot/parsley family
Assaying variation in mitochondrial or chloroplast DNA
Sequence a gene region or the whole mitogenome (DNA sequencing)
Restriction enzyme analysis of a known polymorphism (genetic marker)
Nuclear DNA
Biparental inheritance
Assay for nuclear DNA variation
DNA sequencing (whole genome sequencing)
Genetic marker approaches
Microsattelites:
Highly variable due to high mutation rate
Multiallelic
Typically neutral (not under selection)
Alleles differ by length of sequence
Made up of sequences of repeated blocks of DNA
Genetic markers - microsatellites and SNPs
SNPs: Single Nucleotide Polymorphisms
Arise from point mutations
Bi-allelic
Abundant in the genome (millions)
Easily identifiable with modern genomic methods
2/6/2025
Twinning
Monozygotic (identical) twins
Arise from one fertilized egg which splits into two embryos
Genetically identical at conception
Always same sex
Complications
Monochorionic diamniotic: twins that share a placenta but are in different amniotic sacs
Twin-to-twin transfusion system: Flow of blood from one twin to another causing severe stress on both babies systems
Monochorionic-monoamniotic: twins that share a placenta and are in the same amniotic sac
Can lead to cord entanglement or conjoined twins
Discordance for a genetic condition: de novo mutations can happen after the embryo splits, leading to one twin with a genetic disorder
Dizygotic (fraternal) twins
Arise when two eggs are ovulated and fertilized
Genetic full siblings
2/13/2025
Cancer
All cancers are genetic BUT cancer is very rarely inherited
only 5-10 percent of all caners are caused by inherited mutations
Mutations arise in somatic cells
Six features of cancer cells
Independence of external growth signals
Insensitivity to external anti-growth signals
Ability to avoid apoptosis and autophagy
Ability to replicate indefinitely
Ability of a mass of such cells to trigger angiogenesis and vascularize
Ability to invade tissues and establish secondary tumors
Important concepts
We have several protective systems
Repair
Apoptosis
Immune response
We need several mutations to convert normal cell to tumor cells
Low probability except
Some mutations enhance cell division
Some mutations affect genome stability
Some mutations reduce immune response
Cancer mutations
Multi-stage evolutionary process
You need something to select for cancer expansion, or something that stops you from getting rid of the cancer cell
Cell cycle dysregulation is the cardinal feature of cancer cells
In G1, RBP1 TP53 and CDKN2 play key roles
Associated with somatic and familial changes in tumor cells
Genes that result in acquired cancer cell
Oncogenes — “gas pedal”
When they are normal, they promote cell proliferation
Gain of function mutations result in forms that are too active
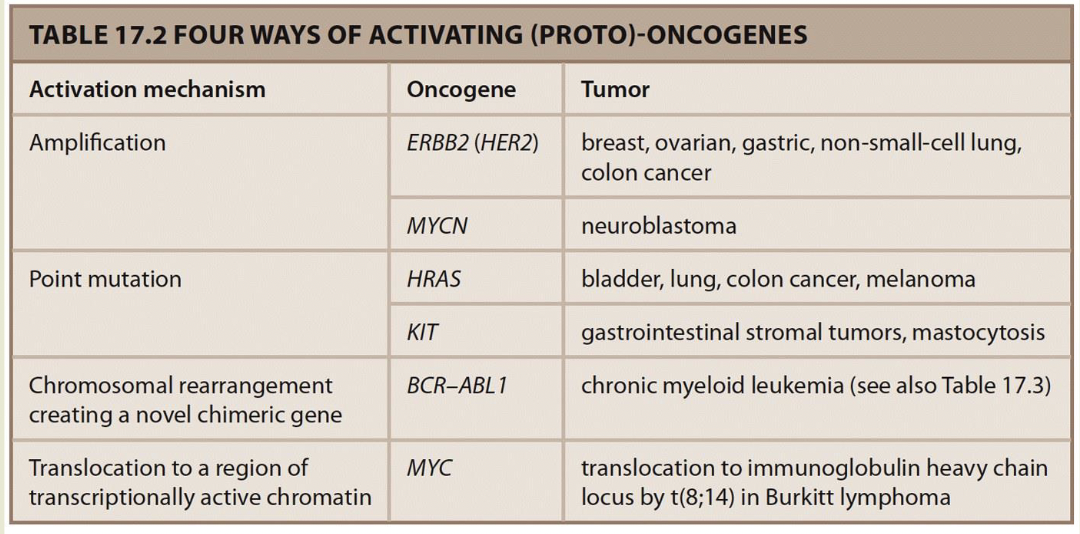
Point mutations decrease activity increasing signal in pathway
GTP-RAS, MAP kinase pathway is on all the time
Rearrangements can activate oncogenes
Philadelphia chromosome: A balanced translocation 9;22 found in 90% of people with chronic myeloid leukemia — BCR-ABL1 chimeric tyrosine kinase fusion protein is always active.
In Burkitt lymphoma 8;14 translocation places MYc in transcriptionally active region - -MYC is upregulated under the influence of the normally highly expressed immunoglobulin genes IGH expresss
Discovered as part of retroviruses capable of transforming cells
Reverse transcribe DNA — form of duplication
Oncogenes turned out to be copies of normal cellular genes (proto-oncogenes).
15% of cancers are caused by retroviral infection
Tumor suppressor genes — “brake”
Normal activity limits cell proliferation, likks incorrigible cells, and maintains genome intgrity
loss-of-function mutations lose ability to control cell proliferation
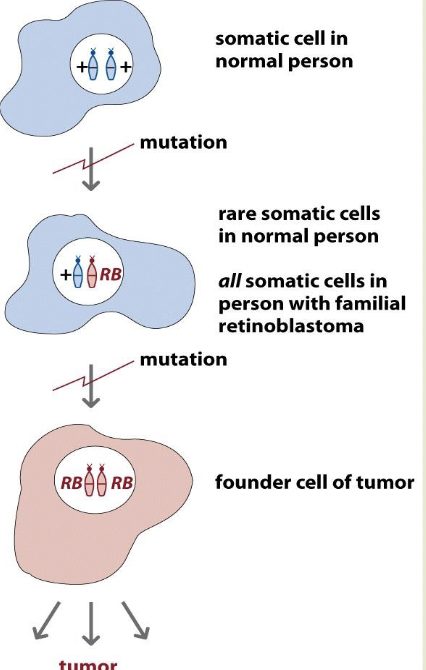
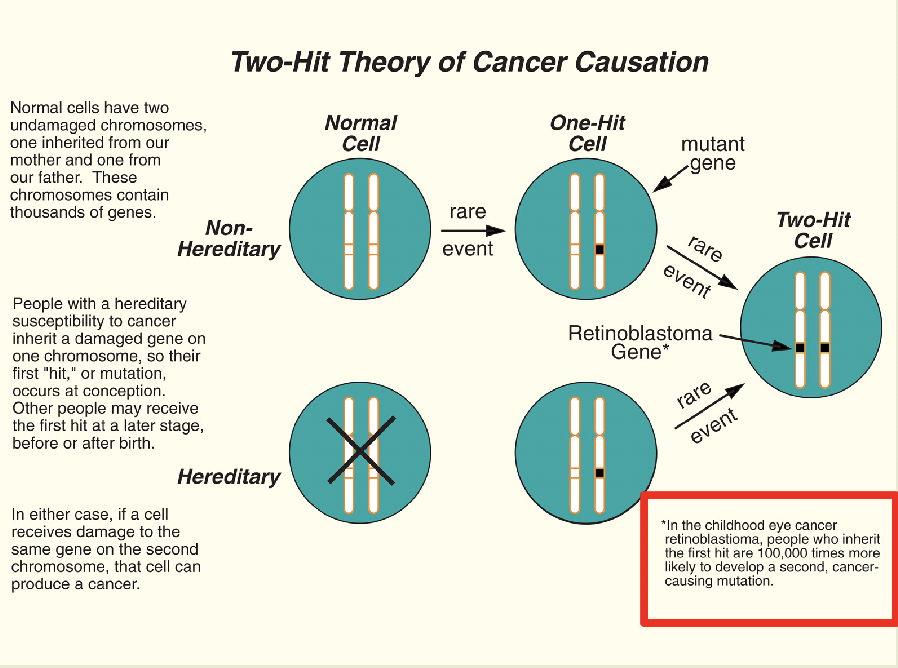
Retinoblastoma is the paradigmatic TS gene and the two-hit hypothesis
You get a tumor when you get a second mutation
Inherited or sporadic mutation
Recessive but behaves like it’s dominant due to two hit hypothesis
Recessive at the cell level, dominant at the retina level
If you inherit the bad allele, you will get the sporadic mutation as well
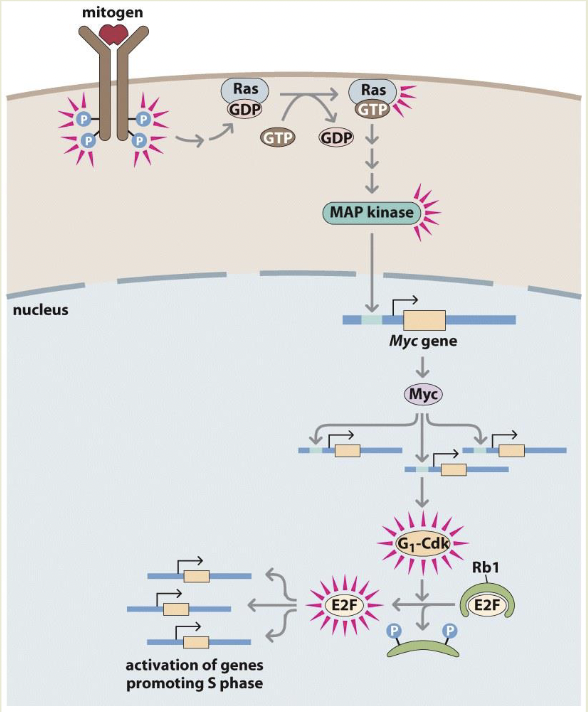
Rb1 binds and inactivates E2F stopping progression into S phase
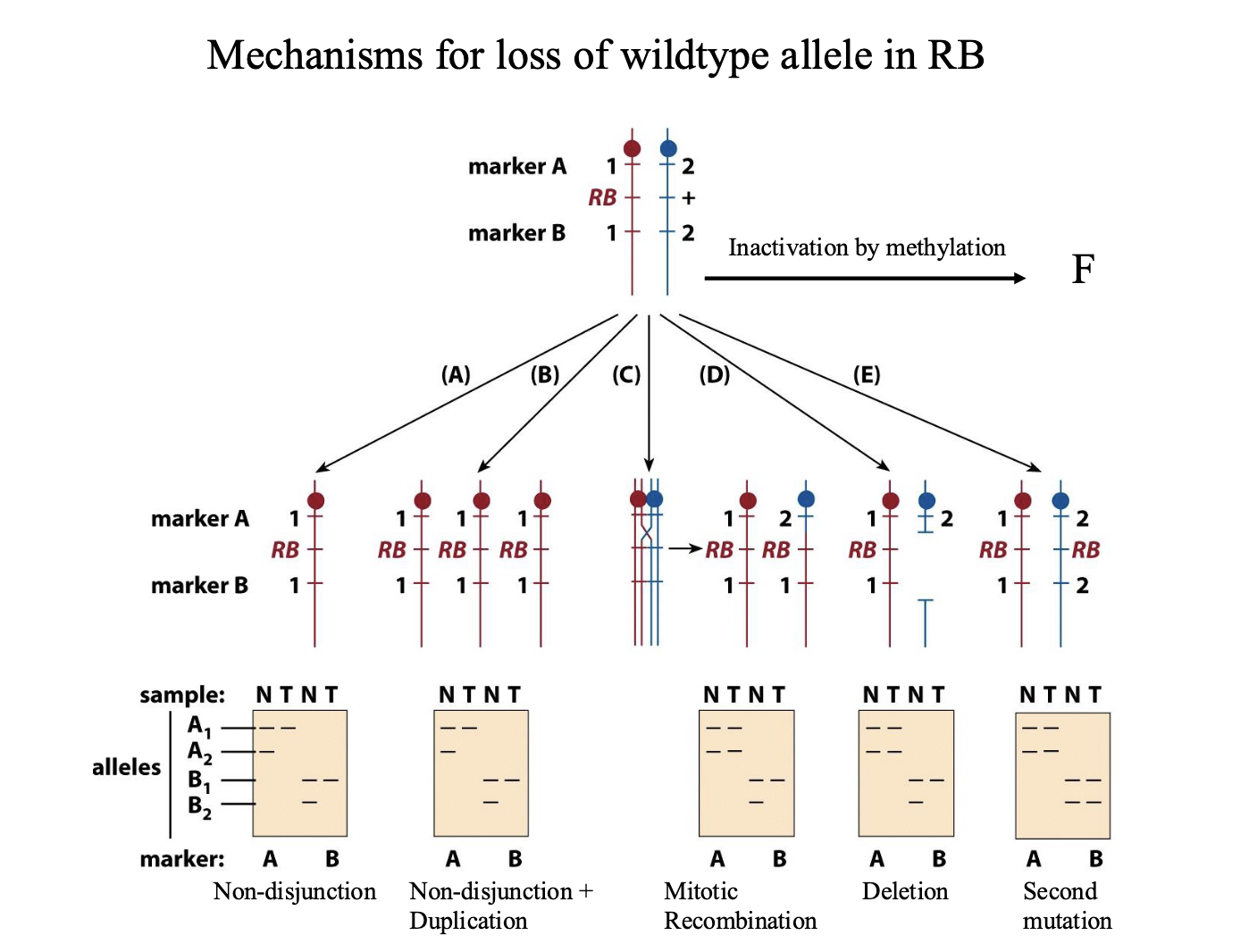
Mechanisms for loss of wildtype allele in RB
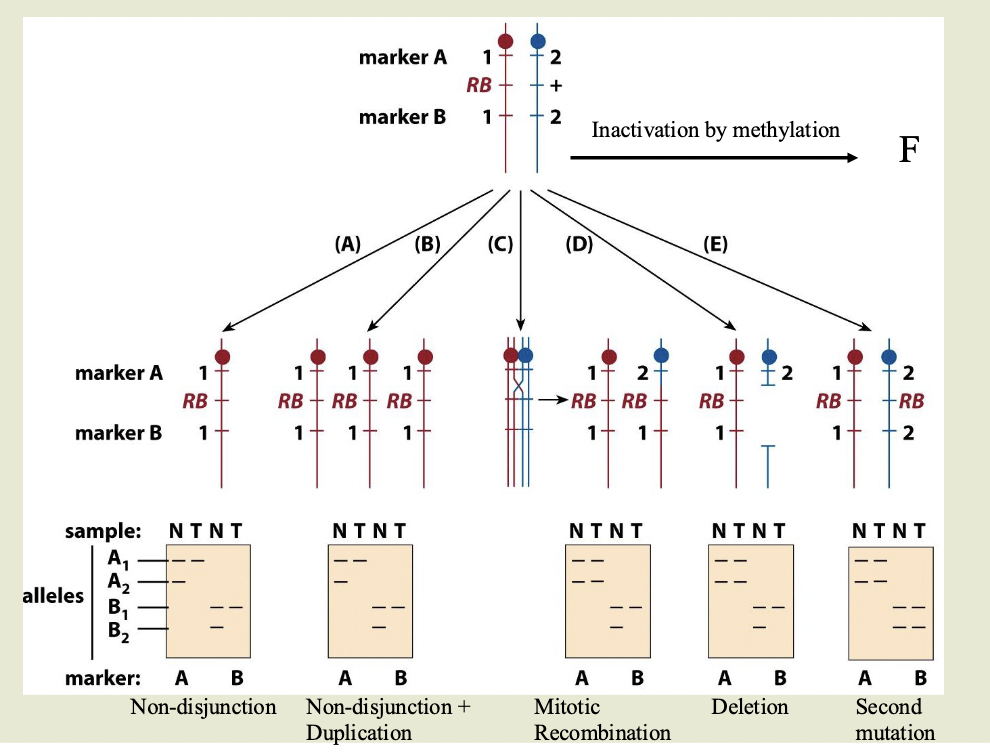
Top of diagram is heritable heterozygous state. 2 markers flank the gene
A1 and A2 and B1 and B2 are different sizes
N=normal. T=tumor
Non-disjunction:
One chromosome with the RB gene, other chromosome is absent
Doesn’t have to be a mutation — this cell is homozygous for RB
Non-disjunction+ duplication
Same as ^ Mutated chromosome is duplicated
Mitotic recombination
Chromosome 2 has recombined and grabbed the part of chromosome 1 with RB
Deletion: delete the part of the wild type chromosome with the allele
Second mutation:
Inactivation by methylation
LOH
LOH= loss of heterozygosity
Used as a marker to locate tumor suppressor genes
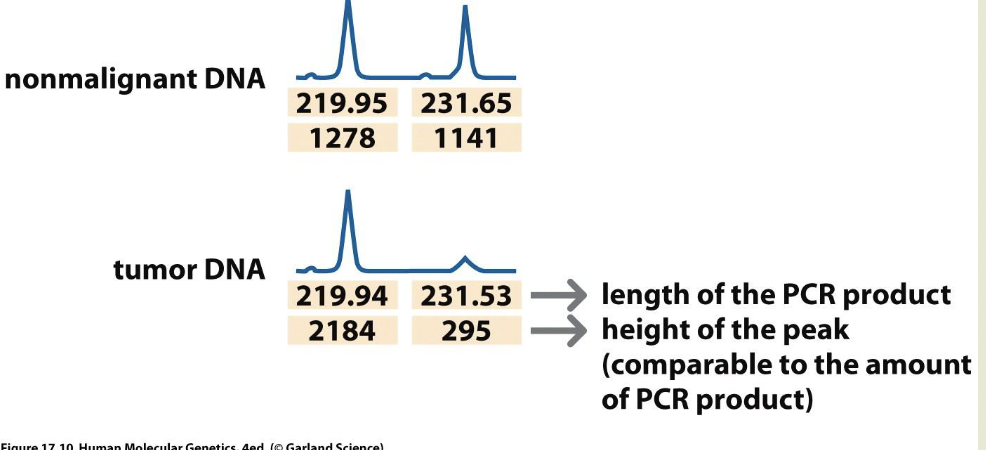
2/18/2025
Cancer facts
77% occurs in people over 55
5 year survival rate is 67%
Cervical cancer
12,000 women per year
Caused by HPV
Strains 16 and 18 most likely to cause cervical cancer
Most common sexually transmitted virus in the US
Virus invades epithelial cells, DNA is integrated into genomes
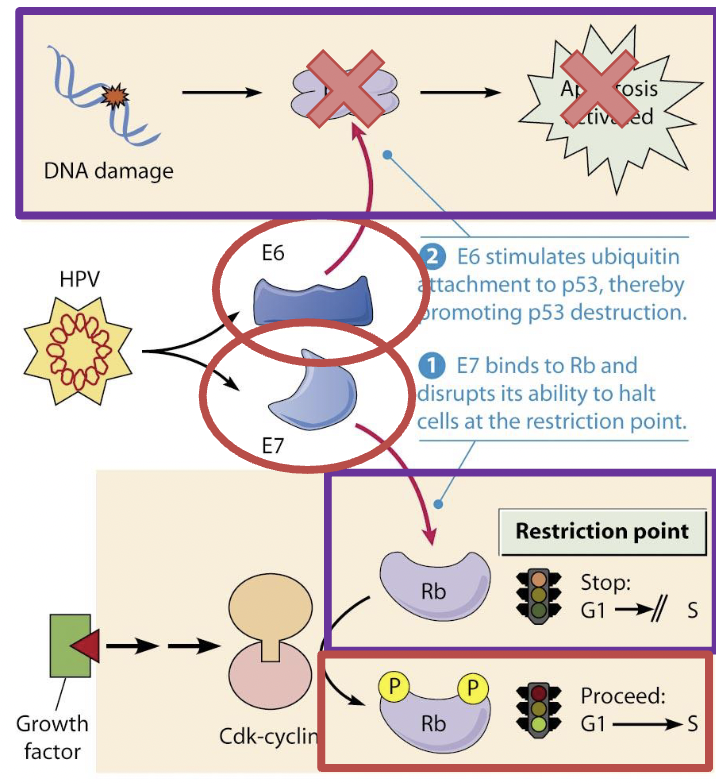
P53 is a tumor suppressor gene
Prevention
Don’t have sex
Cervical cancer vaccine
The philidalpihia chromosome
Chromosomal translocation that generates a kinase involved in the signal transduction cascade that is always on
t(9;22)(q34;q11)
Involved in leukemia
Somatic (not germline) event
Cannot be inherited
3 clinically important isoforms associated with different cancer types
Acute lymphoblastic leukemia (ALL)
Chronic myeloid leukemia (CML)
Chronic neutrophilic leukemia (CNL)
BCR-Abl fusion protein
Tyrosine kinase always on
Increased speed of cell division
Inhibits DNA repair (causing genomic instability)
Cleevec
Treatment for the Philadelphia chromosome
Tyrosine kinase inhibitor — targets BCR-Abl protein
Doesn’t affect other tyrosine kinases
Only effective in Ph-positive CML
Inherited cancers
5-10% of cancer is inherited
Young age of onset
Organs affected bilaterally
Multiple family members affected
Multiple primary cancers
Primary cancer: Arose as the result of a new event in the affected tissue
Recurrent/Secondary cancer: The result of the incomplete eradication or metastatic of another cancer
Retinoblastoma
Unilateral cases have an average onset of 24 months
Bilateral cases have an average onset of 15 months
Inherited retinoblastoma
RB1 gene
Tumor suppressor gene
Up to 8% will have a deletion that could also cause intellectual disability and birth defects
Autosomal dominant at the level of the organism, autosomal recessive at the level of the cell
95% survival rate with treatment
Other cancers can also be associated
Breast/ovarian cancer
Women have a 12% risk of breast cancer, 1.4% ovarian
Inherited breast-ovarian cancer incidence: 1/800-1/2500
5-10% are caused by germline mutations (inherited) in BRACA1 and BRACA2
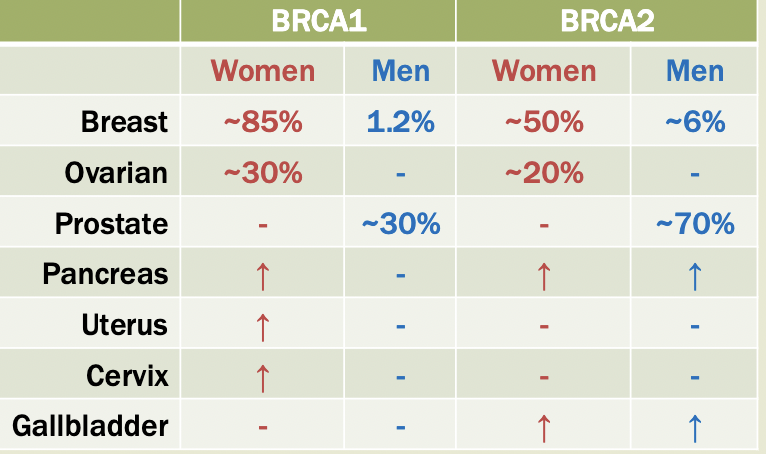
BRCA
Expressed across many cell types
Mutations cause loss of gene function —> tomorrow suppressor genes
Suspected involvement in DNA repair
Management
Climical breast exam
Mammography
Breast MRI
Ultrasound (ovarian)
Blood test (ovarian)
Colorectal cancer
5% risl
Up to 33%B of colorectal cancers are caused by germline mutation but only 3% are highly penetrant
Lynch syndrome: Caused by germline mismatch repair gene mutations
Immunohistochemistry (ICH): Tissue staining procedure, detects the presence or absence of proteins produced by mismatch repair genes
Microsatellite instability testing (MSI): Evaluation for instability in the size of microsatellite markers. More accurate than IHC

Li-Fraumeni syndrome
“huntingtons disease of cancer”
Incidence as high as 1/5000
Caused by TP53 tumor suppressor gene
Cuases many different cancer types
50% of affected individuals will have cancer diagnosed before 30 years old
Multiple endocrine neoplasia
MEN1: Wermer’s syndrome
MEN2A: Sipple syndrome
Incidence 1 in 35000
Autosomal dominant
RET gene, 10q11.2
Protooncogene
Regulates cellular environmental responses, promotes growth and division
Mutations over-activate the gene (tyrosine kinase is always active)
RET gene mutations also cause Hirschprung disease
MEN2B:
Liquid biopsy
Used to test fetal genome from mother’s blood
Used for tumor diagnosis — tumors have genomic instability
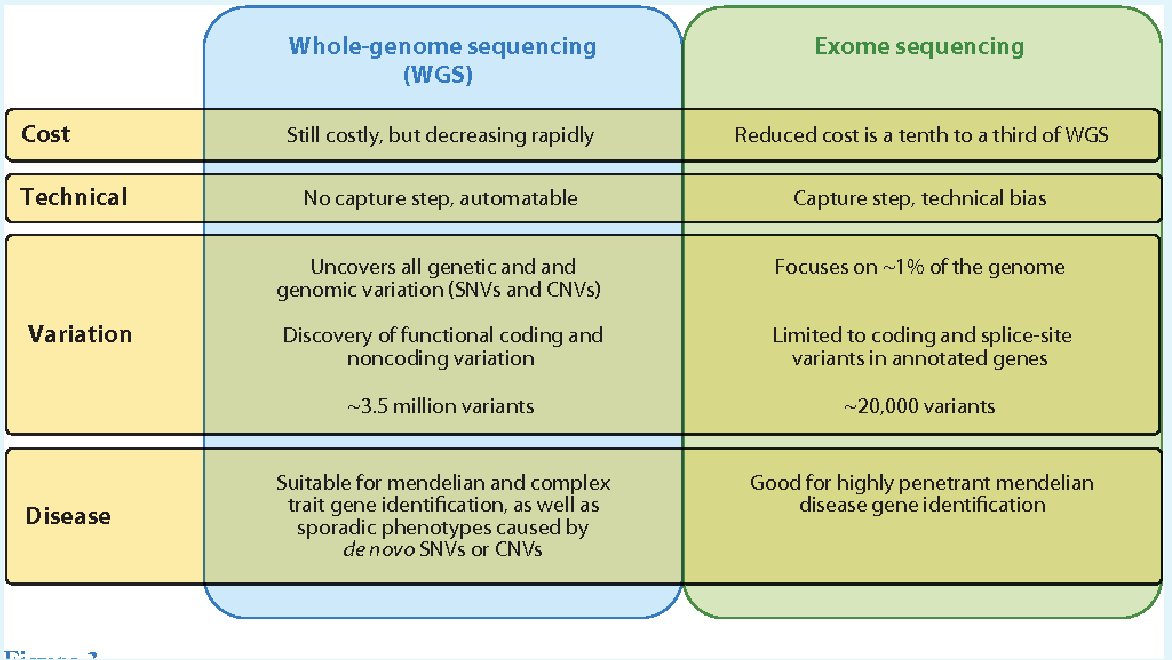
2/25/2025 Analyzing genomes using technologies
testing
Diagnostic testing: confirms or rules out specific genetic condition
Predictive and pre-symptomatic testing: used to detect mutations associated with disorders that appear after birth but before symptoms (ex: BRCA)
Carrier testing: used to identify individuals who carry one copy of a recessive condition
Preimplantation genetic diagnosis (PGD): used in in vitro fertilization to identify embryos without genetic abnormalities for implantation and pregnancy
Prenatal testing: Used to detect changes in a fetus’s genes or chromosomes before birth (amniocentesis or chorionic villus sampling)
Newborn screening: Testing newborns for disorders (PKU or phenylketonuria)
Testing methods
PCR: makes millions of copies of a target DNA sequence from very small sample and is the base technique for most genetic testing
Key techniques in most genetic tests
YOU WILL BE ASKED TO DESIGN A PCR EXPERIMENT ON THE EXAM
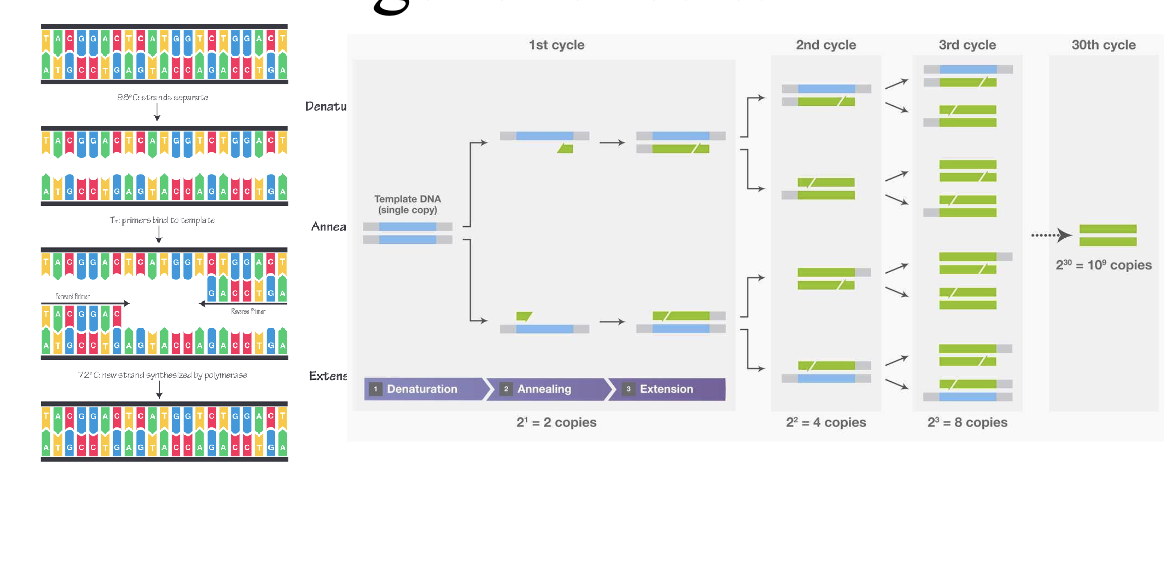
Directions are important (replication goes in 5’ to 3’ direction)
New bases get added to 3’OH
Next generation DNA sequence: Sequencing of an individuals entire genome
Microarrays (DNA chip technology: Analyze gene expression and detect SNPs. Basis of 23 and me
Karyotyping: Visual examination of chromosome number and structure
FISH: uses fluorescent probes to target specific sequences to assay for specific sequence
Biochemical genetic testing: examines the amount or activity level of proteins that can indicate abnormalities
Types of variation
SNPs
Deletions and duplications
Indel: deletion and duplication <1000 bases
CNV: deletion and duplication >1000 bases
Trinucleotide repeat expansion (TNR): increased number of trinucleotide repeats in a given area of a gene (Friedeich ataxia, Huntington
Methylation: Addition of methyl groups to DNA can turn the gene off
Mitochondrial DNA variants (MtDNA). Changes to the DNA in the mitochondria
Clinical genet tests
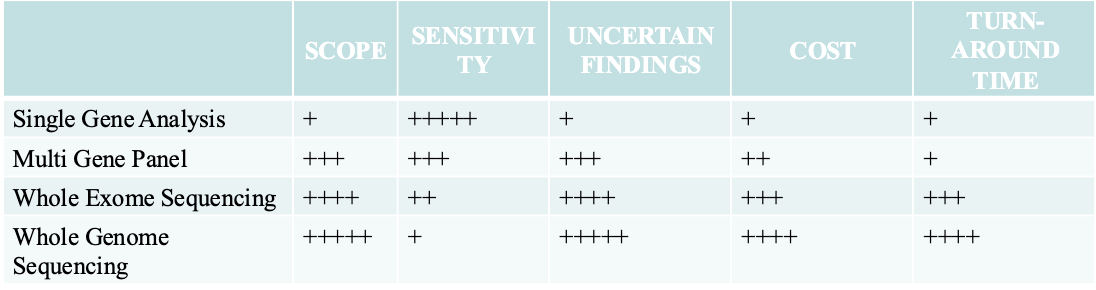
Single gene testing: Single gene testing is done when there are symptoms of a
specific condition or syndrome. (i.e. Duchene muscular dystrophy or sickle cell
disease) or when there is a known genetic mutation in a family.
Panel testing: Looks for changes in many genes in one test and are usually
grouped in categories based on different kinds of medical concerns (i.e. low
muscle tone, short stature, or epilepsy or risk of developing breast or colorectal
(colon) cancer
Large-scale genetic or genomic testing. There are two different kinds of large-
scale genetic tests.
Microarrays and SNP arrays looks for INDELs and predetermined SNP
Whole Exome sequencing looks at all the expressed parts of genes in the DNA.
Whole Genome sequencing looks at all of a person’s DNA, not just the genes.
Large scale genotyping in humans
Screening
DNA microarrays
Focused genome sequencing
SNP chips
PCR amplification
Hybridization capture
WGS
Sxperimenting
Transcription profiling
RNA arrays, RNA seq, qPCR, ddPCR
Epigenetic analysis
Protein DNA interactions (ChIP-seq)
Human genetic variation
Chromosome variation: typically limited to larger deletions, duplication. and translocations 5Ml>
Segmental values (copied number variation): 1kb-5kb
Most but not all are associated with segmental duplications
Responsible for most differences (per base tail) between humans
Single nucleotide polymorphisms (SNPs)
Base substitutions and small Indels
COULD BE ON EXAM

Microsatellites (SSRs)
Several short repeated motifs
Highly polymorphic
Good genetic marker
Good forensic tool
Sometimes directly related to disease
Huntingtinon’s and fragile x
Assay by PCR and length analysis
Not all trinucleotides
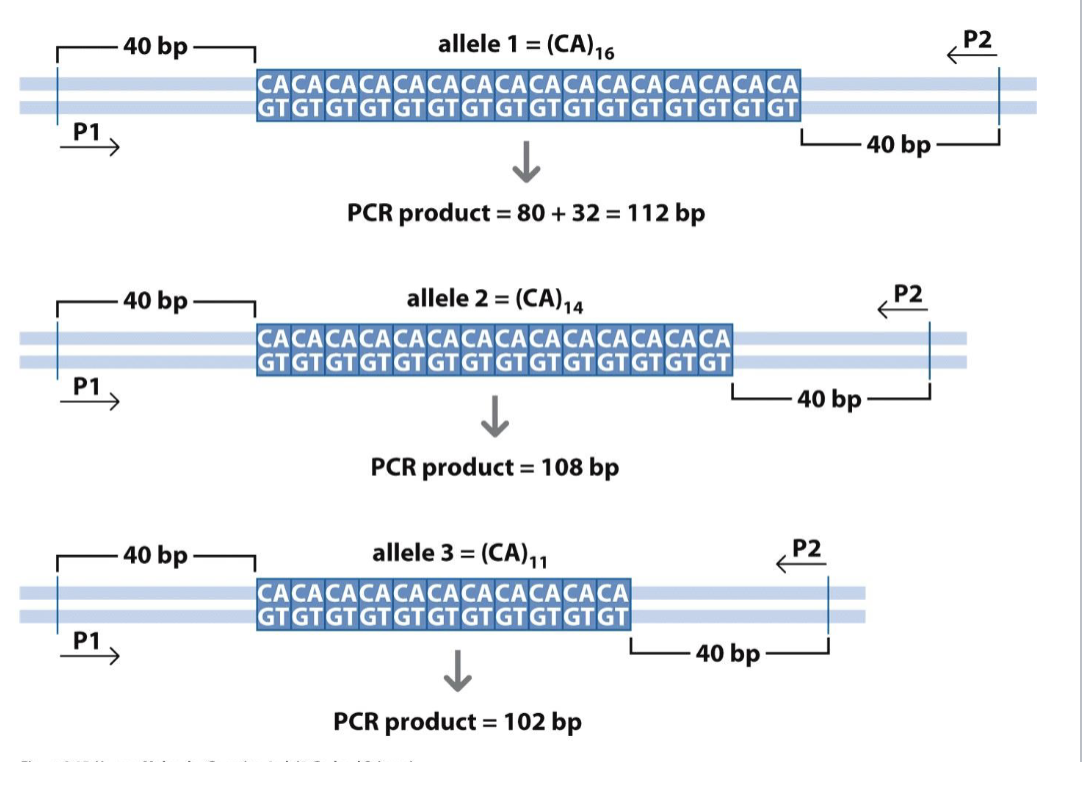
Different lengths=different alleles
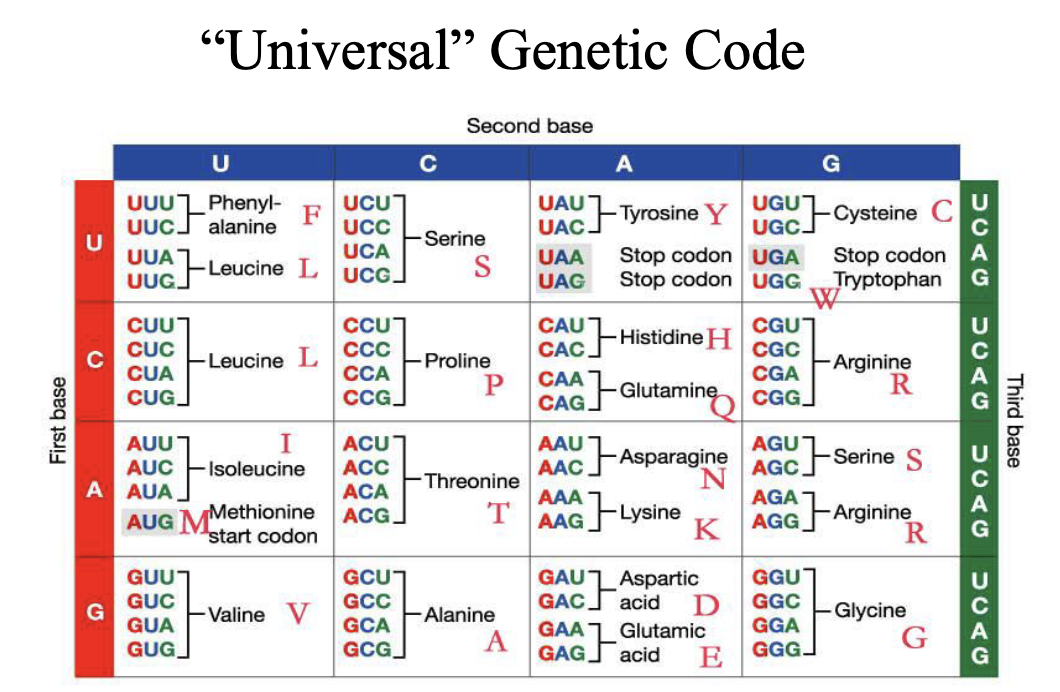
PCR
How do we identify and detect a specific gene sequence in a genome?
2 big issues
There are a lot of other sequences in a genome that we’re not interested in detecting (specificity)
1/4n is the probability of finding a sequence at random
The amount of DNA in samples we’re interested in is very small (amplification). Contamination is very dramatic
Type of question that will be on the midterm
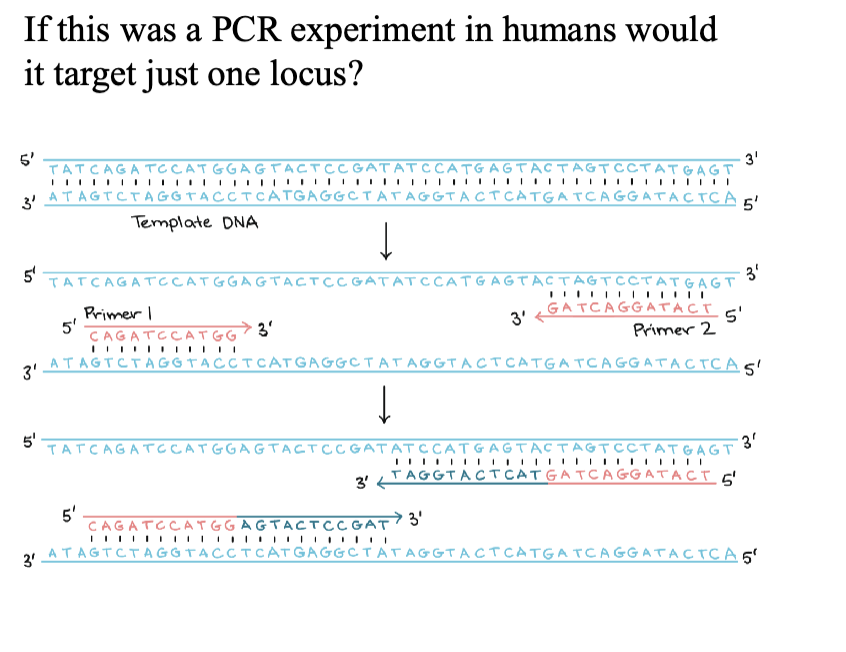
Mark the orientation of the forward and reverse primer
2/28/2025
exam question: match red and blue lines with the PCR
Testing
Microarrays are useful for detecting DNA deletions and duplications
Sequencing
Traditional DNA sequencing/chain termination quequencing/sanger sequencing
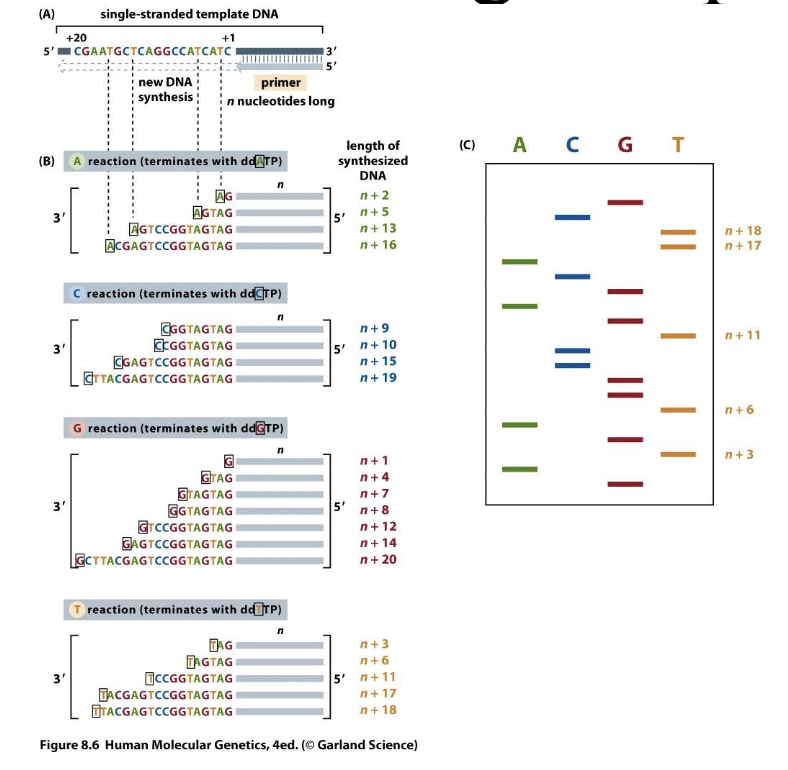
Separate strands of different lengths by gel electrophoresis
Used dideoxyribonucleic acid — H on the 3’ carbon means it won’t bond with anything else
Next generation DNA sequencing technologies
Much cheaper — don’t have to run gels
It is still expensive to analyze the genome
Sequencing by Synthesis (Illumina)
Second generation sequencing
Goal is to monitor replication
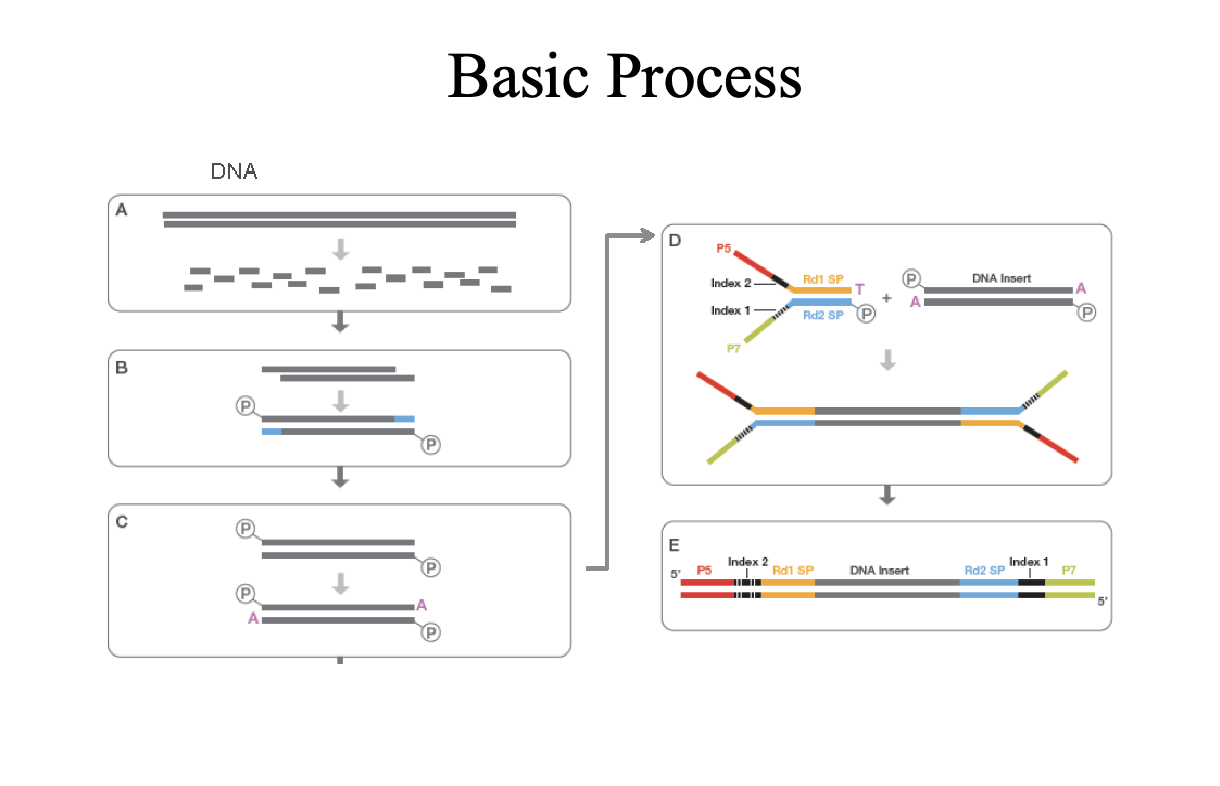
P5 and P7 (red and green) are primers sites
Sequence on the inside surface of a flowcell
Primers are covalently attached to the glass
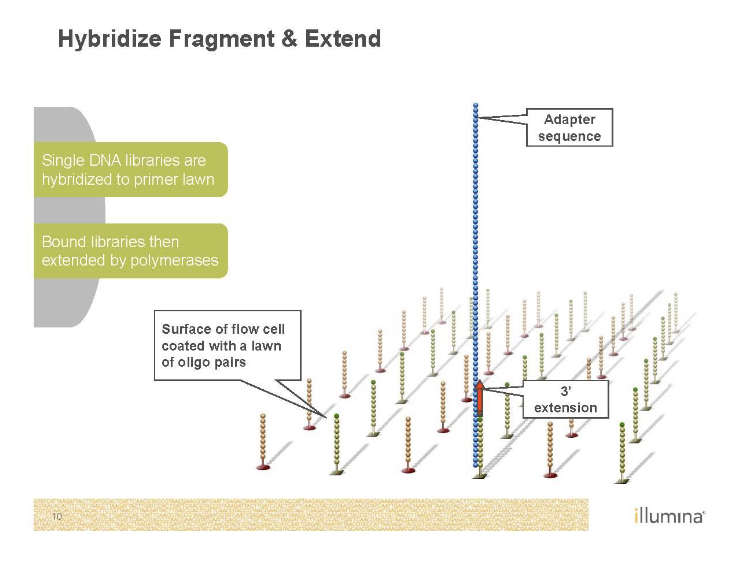
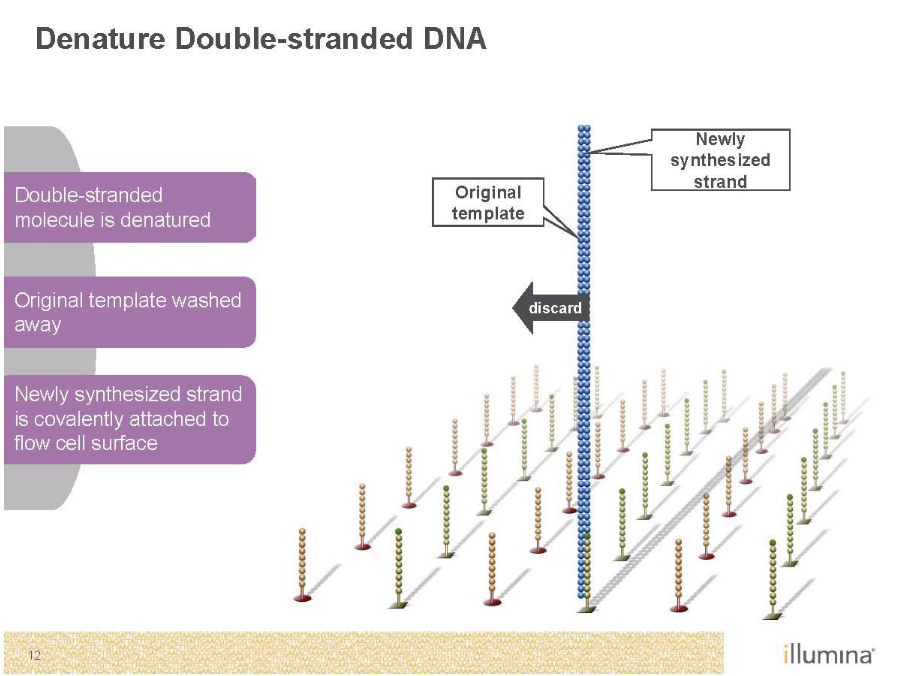
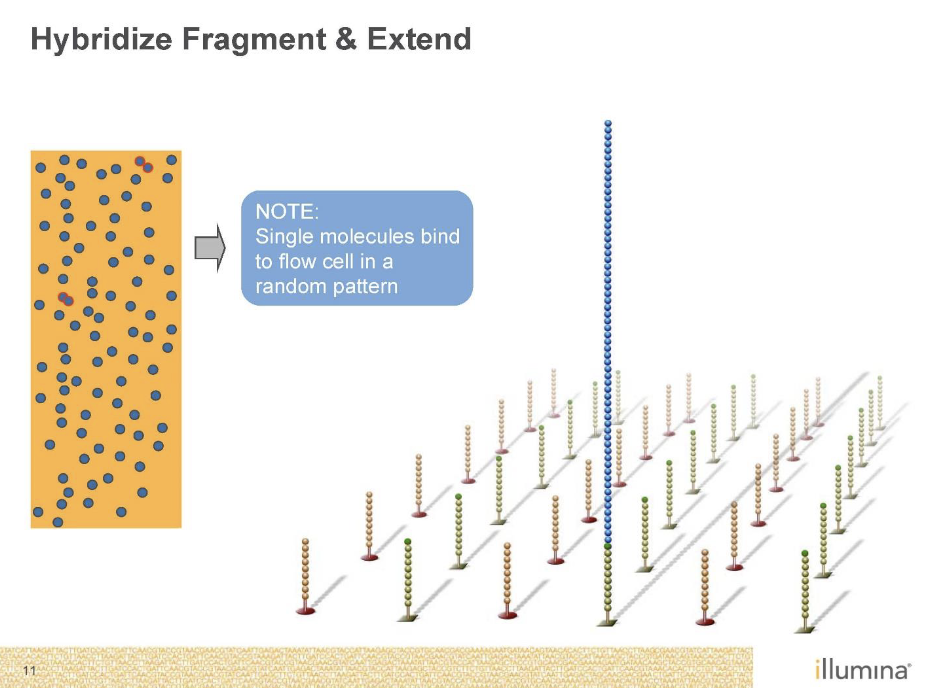
Needs to be amplified with PCR as well’
Bridge PCR
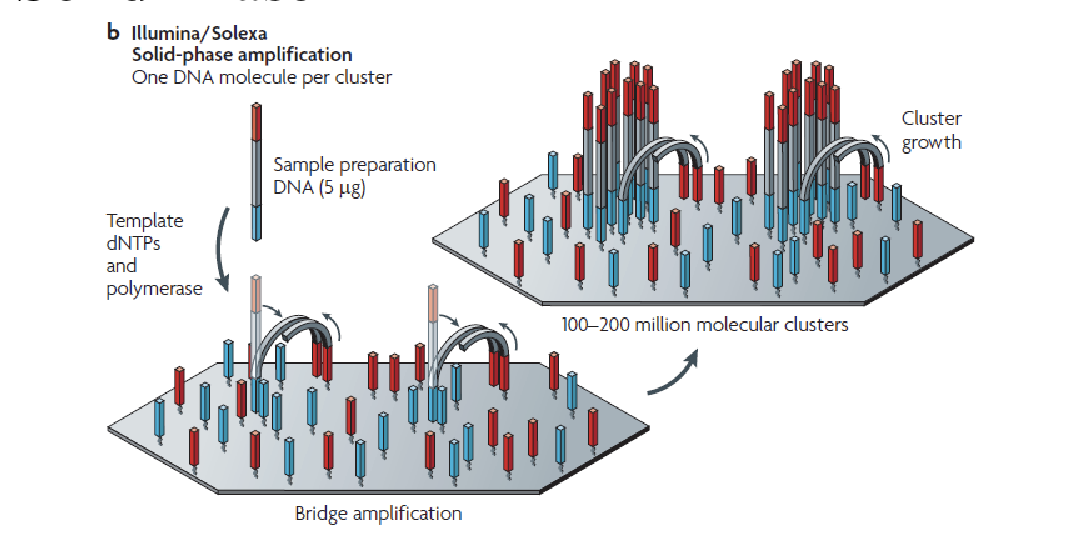
Just like normal PCR except primers are stuck to the glass
End up with clusters of identical DNA molecules
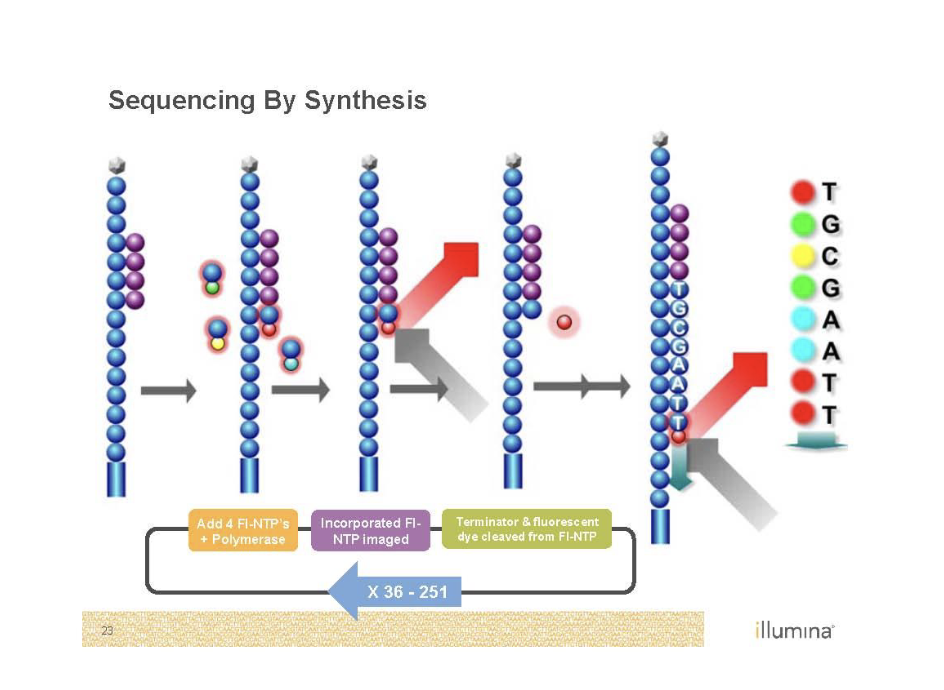
Need to get rid of color
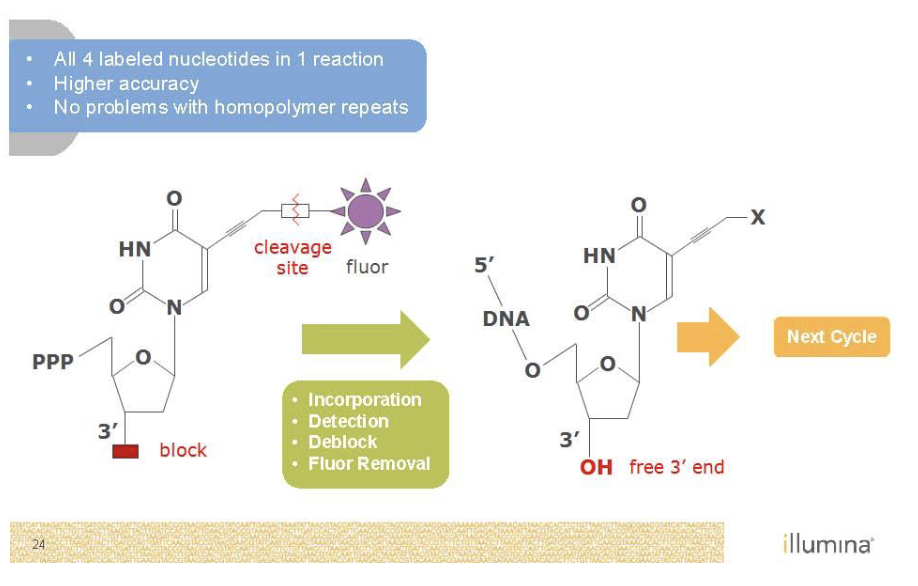
Illumina stargazing
PacBio
A bunch of wells in a plate
At the bottom of the wells there is a hole
Holes are such a size that we can look in but nothing will come out
Incorporating fluorescent nucleotides
Single molecule real time sequencing (SMRT)
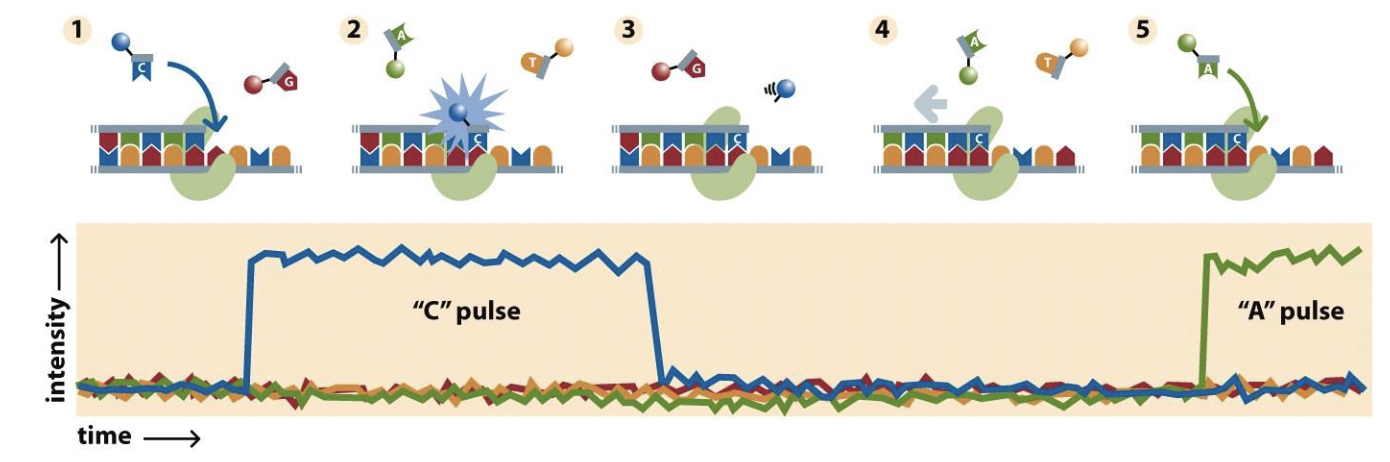
Nanopore sequencing
You have a protein hat unzips the DNA and pumps the sing strand through a pore in a membrane
Measure change in electrical activity across the membrane at every pore
Electrical activity changes for each nucleotide
Really long reads
MinION field sequencer
KNOW THE 4 SEQUENCING TECHNOLOGIES
Array based genotyping
Arrays contain oligonucleotides that test the seuqnce of a sample
Arrays target the test to a set of loci
They function based on the discrimination of complementary base pairing or the ability to extend a base at a specific location
Transcription profiling
Arrays can be used to measure the amount of RNA produced by a set of genes
Sequencing can be used to measure expression of a genome
Epigenetics
Methylation — histones influence what portions of the genome are available for transcription
bisulphite sequencing
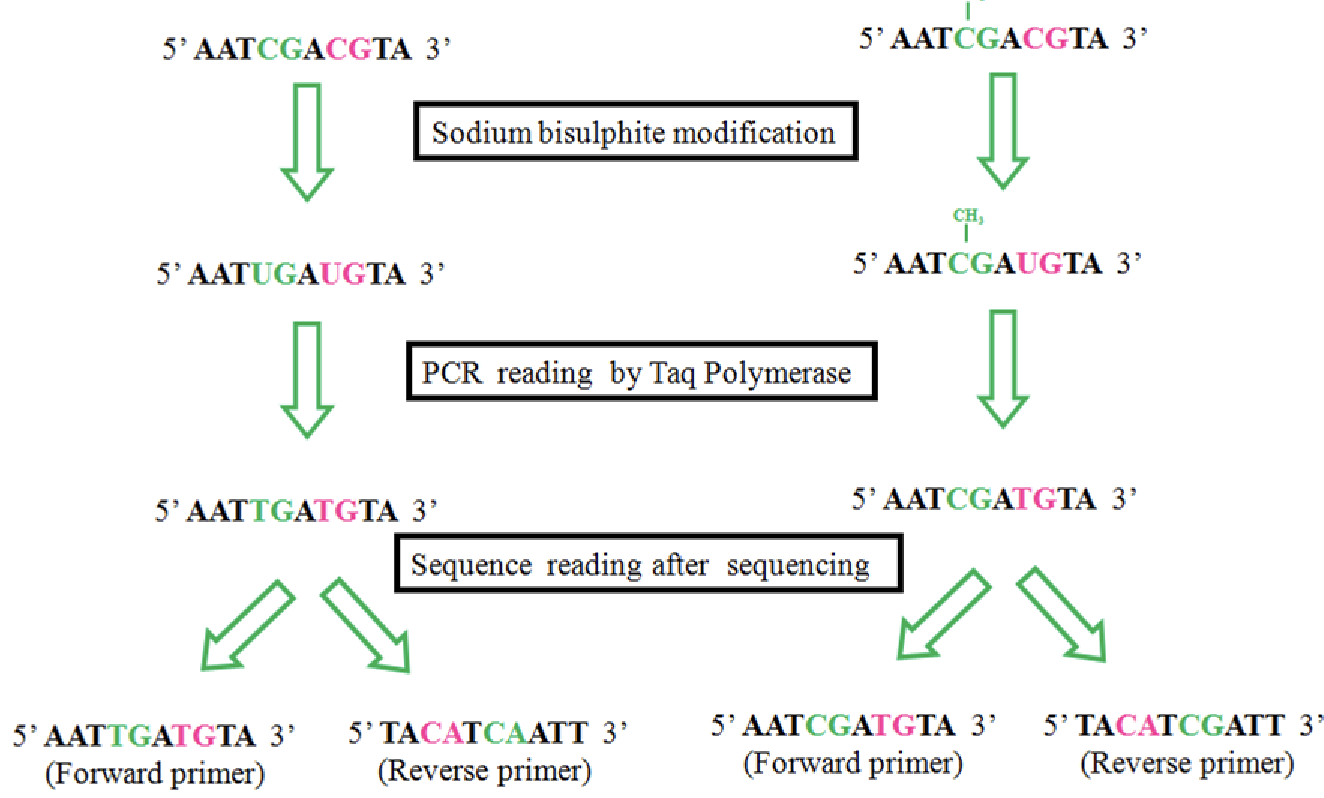
If the C is not methylated, it switches it to be a U
Everywhere C that is remaining you know is methylated, so you can map these points
ChiP-seq
Used to study protein DNA interactions
3/4/2025
Standard of care
Standard of care: that which a minimally competent physician in the same field would do under similar circumstances.
Not intended to replace the good judgement of clinicians
Cell free fetal DNA testing
Can be used to detect gender as early as 8 weeks
Female is more susceptible to error
Takes blood sample from mom
Karyotyping vs array
Both detect aneuploidy and segmental variation
Microarray can detect much smaller CNVs than karyotype
Karyotype can detect balanced translocations (microarray can’t)
Case 1
You are as ked to evaluate Sally, 2 years old, for a history of Tetralogy of Fallot, clubfoot (bilateral), failure to thrive, and recurrent infections. She is otherwise healthy and has shown normal development. Her parents report the following family history.
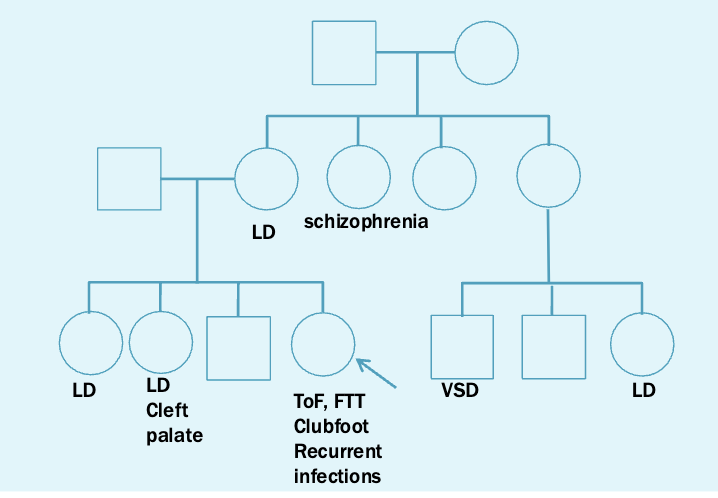
Teralogy of fallot
Heart defect
Strongly associated with 22q11.2
6% of isolated cases of ToF have 22q11.2 deletions
22q.11.2 deletion syndrome: digeorge syndrome
Incidence 1/4000
Can test using karyotyping or FISH or microarray, but microarray is standard of care
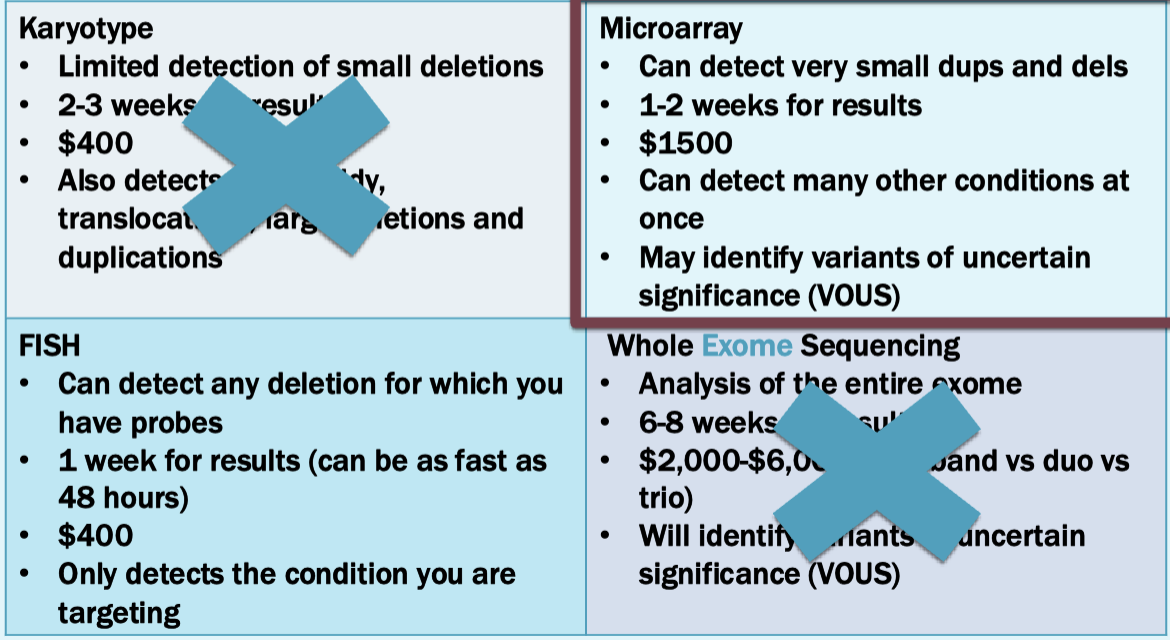
For sally, the microarray identifies a 22q11.2 deletion
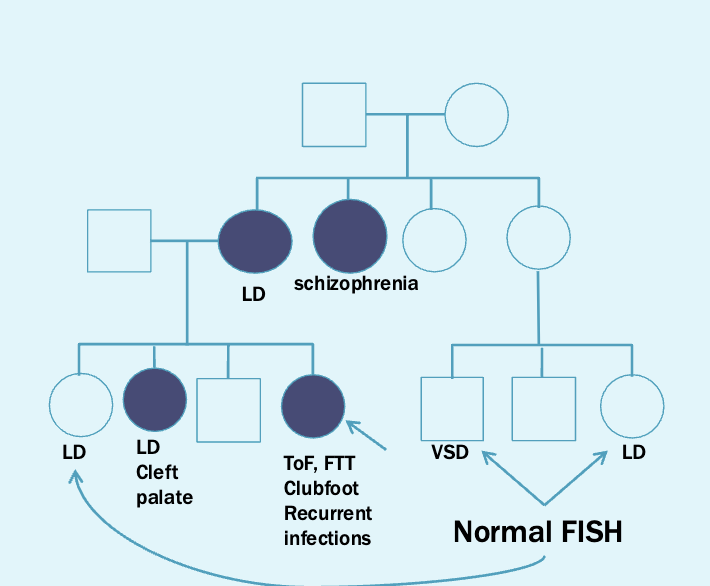
Next step is to offer testing for other family members via FISH
Case 2
Betty is 19 weeks pregnant and has been referred to your practice. Her obstetrician is concerned because a recent ultrasound showed very small nasal bone, small cerebellum, choroid plexus cyst, and a single umbilical artery. Your ultrasound confirms the fetal findings. Betty and her husband, Luke, report that there is no significant family history
Small nasal mone — Down
Choroid plexus cyst — Trusomy 18
Single umbilical artery — could be anything
Options

Microarray is best option
A 5p15.2-p15.33 deletion is detected: Cri-du-chat (cat cr y) syndrome
1/20000 -1/50000 incidence
Newborns make cat like cry
Microcephaly
Severe intellectual and motor disabilities
Dysmorphic facial features
Next step: find a critical region
We know this is a spontaneous event

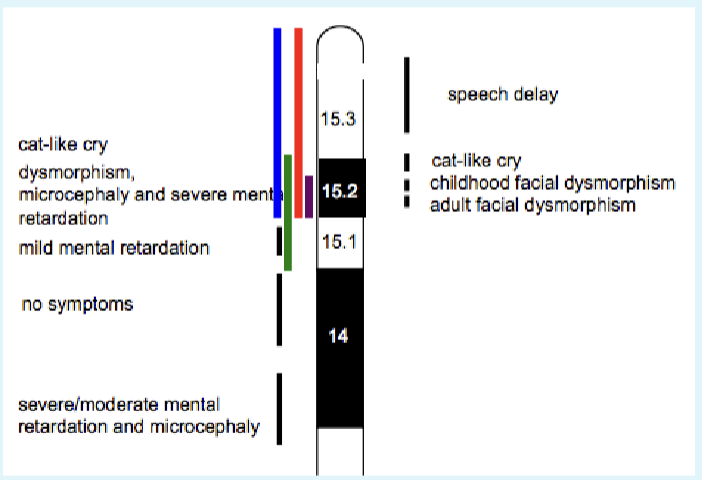
Look at where deletions actually span. Places critical region for cat-like cry phenotype in the 15.2 area
Case 3
Edward is a 7 year old boy with a history of microcephaly, atrial septal defect, long palpebral fissures, large ears, polydactyly, intellectual disability, elevated liver enzymes, mild anemia, and several food allergies including eggs and
wheat. He has already had an extensive genetic work -up which as been normal thus far. You are seeing Edward and his mother, Rhonda, for follow-up
Tests already done
Karyotype: 46,XY
Fragile X syndrome: normal
MECP2 analysis (atypical Rett syndrome): normal
L1CAM analysis (X-linked hydrocephalus): normal
Williams syndrome: normal
Mitochondrial function studies : normal
Microarray: normal
Options

Whole exome sequencing is the best option here
The Rominovs
Determining who was in the mass grave
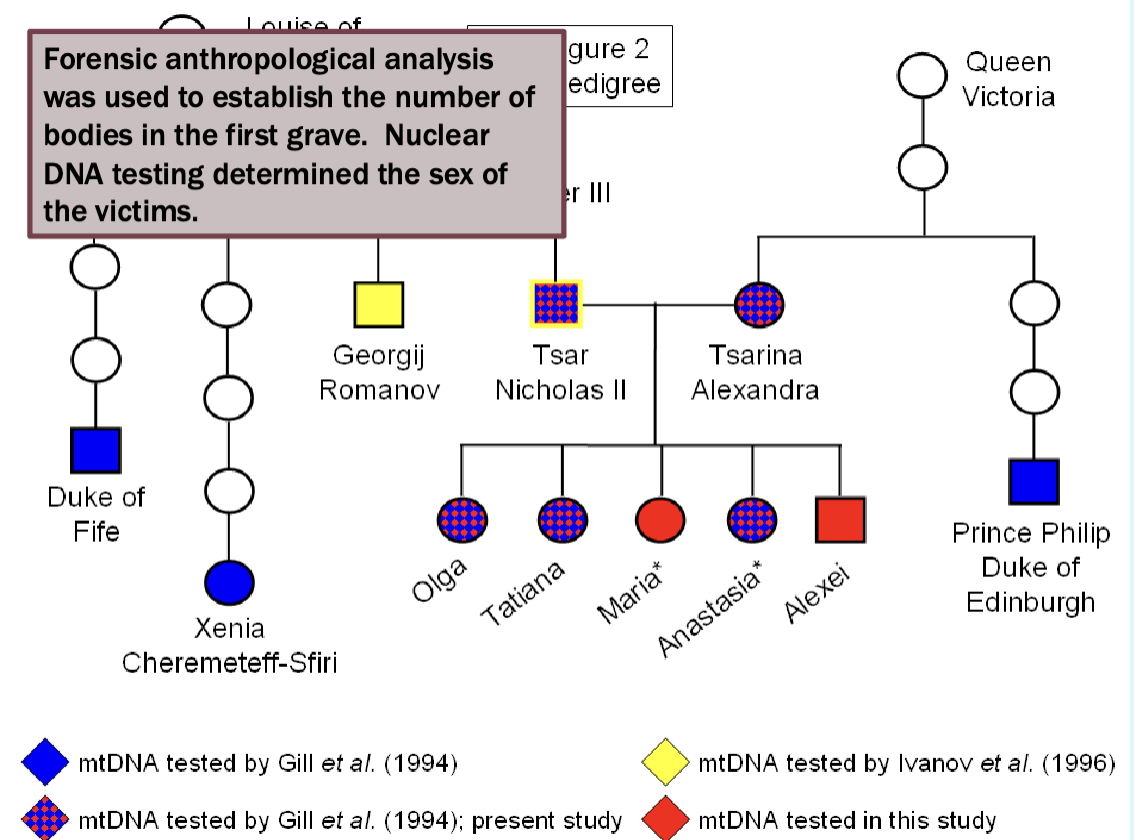
Focused on mitochondrial DNA
We understand it’s the inheritance patterns
Evolves rapidly
Mitochondrial DNA testing showed that Prince Philip shared a common female ancestor with Alexandra and three of her daughters from the first grave
Also showed that Duke of Fife and Princess Xenia shared a common female
ancestor with Nicholas.
Nicholas’ remains showed a single point heteroplasmy in the mtDNA (16169 C/T) that was not seen in his maternal relatives (16169 T). This rare
heteroplasmy was shared by his brother, Grand Duke Georgij Romanov.
But
The remains of only 3 children were discovered in the first grace, Alexei and one sister were missing
Found the second grave in 2007
The second grave
Forensic anthropological analysis
The second grave contained two people
15-19 year old female
12-15 year old male
Ethnicity and height could not be determined for either victim
Silver fillings showed that at least one victim was an aristocrat
The grave was over 60 years old
DNA analysis
DNA was extracted from several bone sites (mostly long bones)
PCR was used to amplify and quantify (real-time PCR - qPCR) the
extracted DNA
Multipex PCR was used to used to amplify and sequence the mt DNA
DNA from the Y chromosome was also amplified using PCR
Samples from the second grave matched the mtDNA studies from the first grave and from prince Philip
Genotyping results
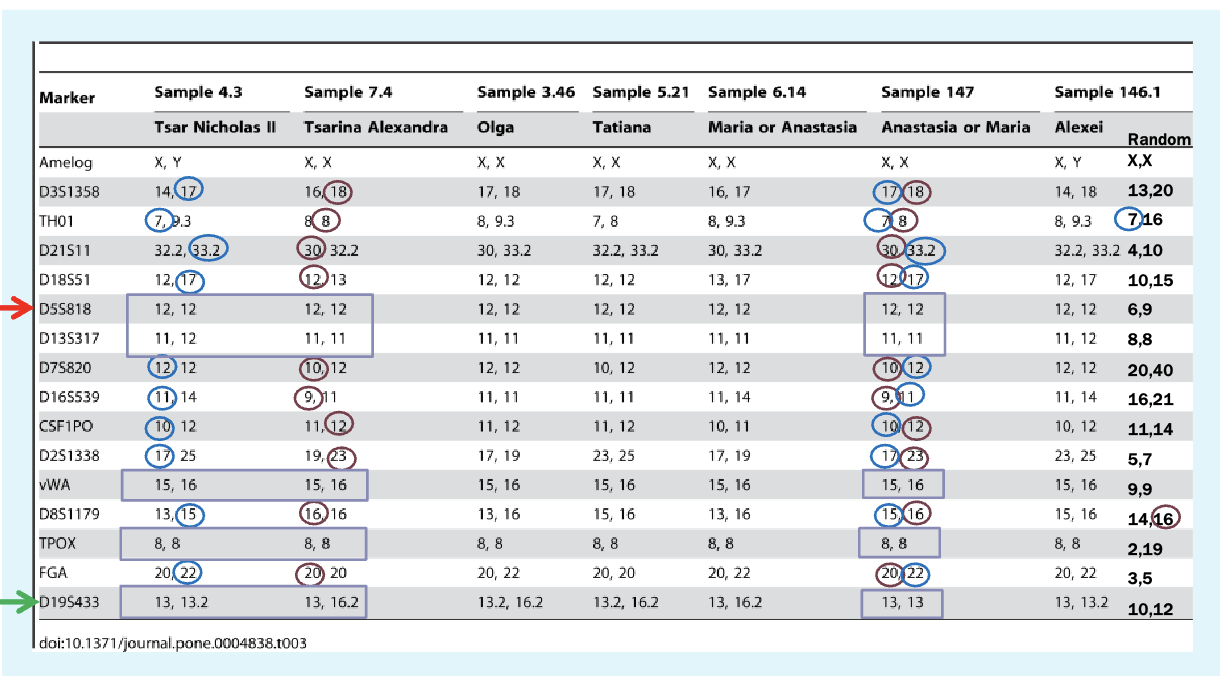
Markers are microsatellites
Found using PCR — design primers to look for specific primers
Y-linked marker results
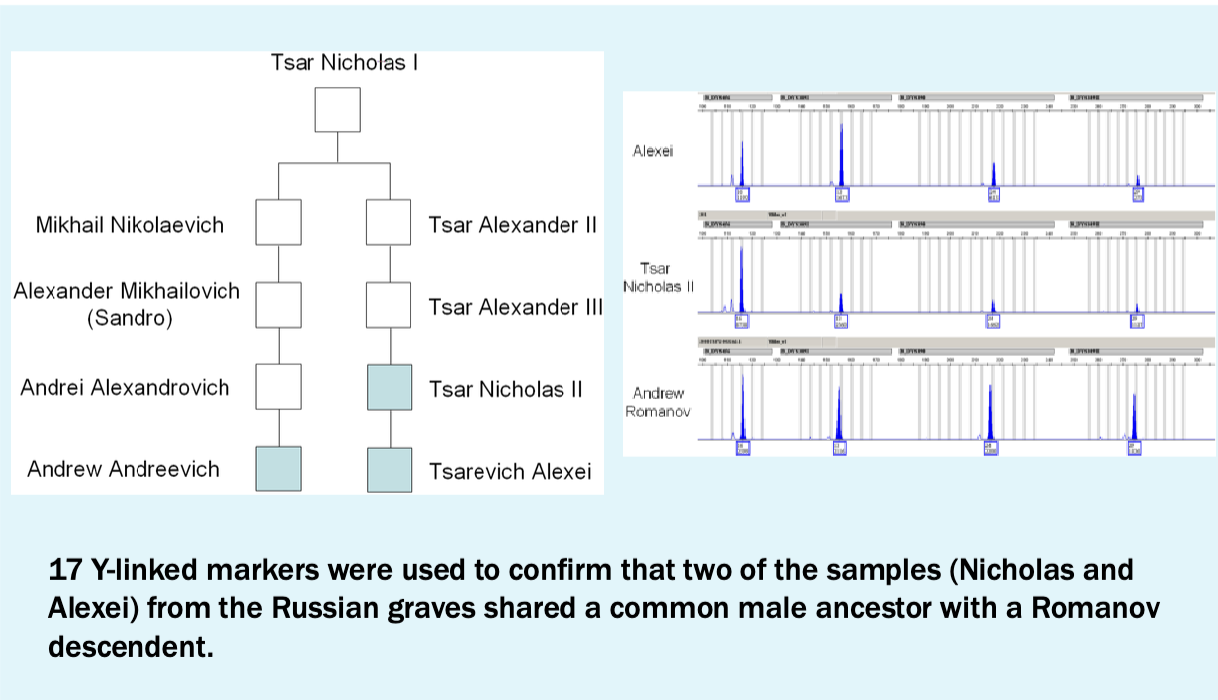
Conclusions
All of the Ramonov family were murdered on July 17, 1918 and buried in two separate mass graves
DNA forensics
CODIS
CODIS: Combined DNA Index System
Operated by the FBI
Contains DNA profiles from convicted offenders and arrestees in states where collecting this data is legal
Contains DNA profiles found at crime scenes
Only works if everyone decides to use the same markers!!
Process
Law enforcement agencies submit DNA profiles derived from samples
taken as part of a criminal investigation
Sample DNA profile is compared to the CODIS database
If a match is found in the Convicted Of fender/Arrestee database, it is usually possible to obtain a warrant to collect a sample from the matched an individual for a reference sample
If a match is found in the Forensic Index the crime can be linked to
one or more other crimes
Accepted DNA profiles
STR (short tandem repeats): 20 loci
Y chromosome STR
Mitochondrial DNA (mtDNA)
Second 2 only ised to identify missing persons
GEDmatch
Online tool
Users submit their raw DNA for analysis and comparisons with other users
Has been used by law enforcement to identify suspects and missing persons
Has a lot of data from both ancestry and 23andme
Open to everyone
The Golden State Killer
Joseph James DeAngelo murdered at least 12 people
Cold case for 44 years
Arrested in 2016 at age 72
No match for DNA sample in CODIS
Comparison of DNA taken from a crime scene was submitted to GEDmatch and several partial matches were made
Degree of relationship to the sample DNA was inferred from the amount of similarity between the sequences — people who he was related to
This provided a narrower field of suspects
Brave new world
Analysis of a DNA sample taken from a crime scene can be used to infer ethnicity and physical characteristics
Modeling software was used to show the effects of age
Paternity testing
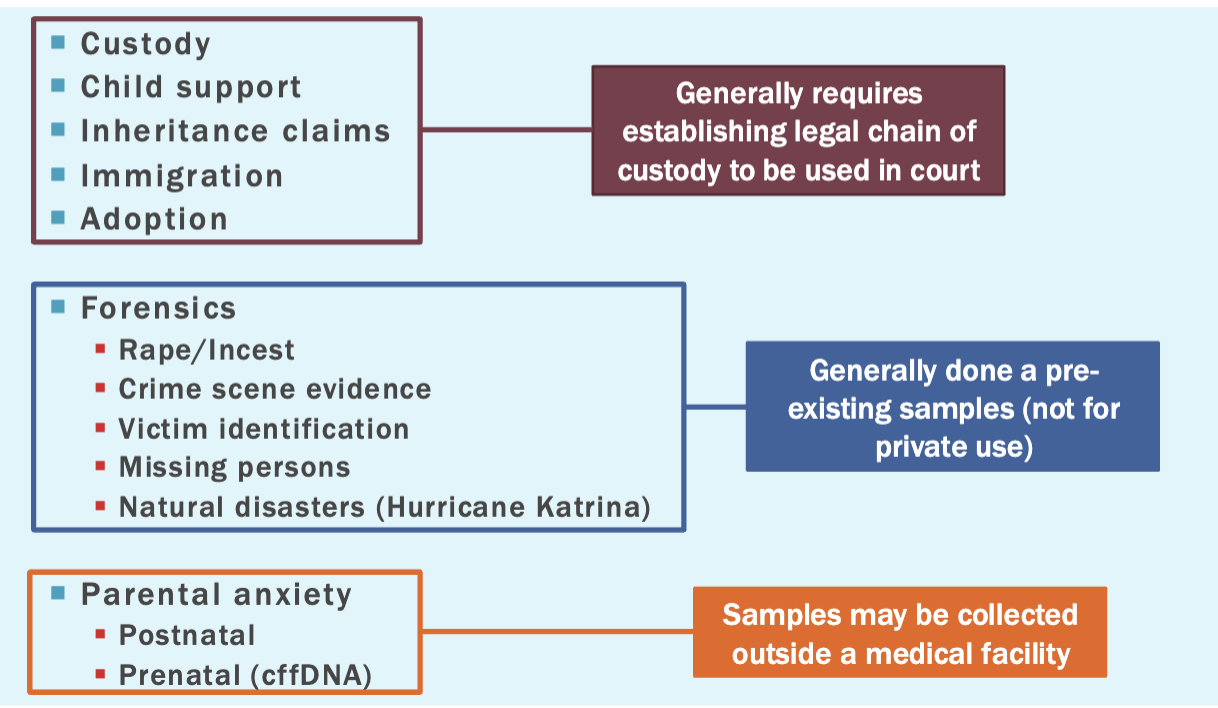
Analysis of 10-20 markers
3/6/2025
Exam info
modules 4 5 and 6
New genes come from gene duplication
Mechanisms of gene duplication
Tandem gene duplication (direct or inverted) unequal crossing over (homologous chromosomes) or unequal sister chromatid exchange (same chromosome)
Duplicative transposition (typically retrotransposition)
Ancestral cell fusion (think endosymbiont theory)
Large-scale, sub genomic duplication (segmental duplication inter and intra chromosomal)
Whole genome duplication
Protein coding genes beget protein coding genes
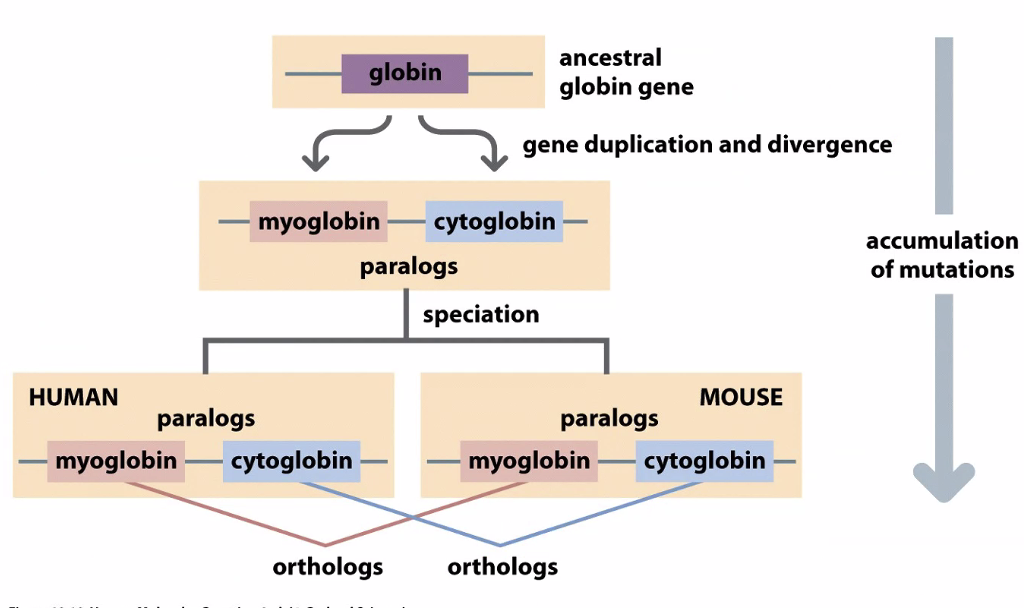
Involved in oxygen transfer
We use phylogenetic trees to understand how genes evolve
Globins diverged in evolution
Usually when there is a gene duplication, one becomes nonfunctional
Neofunctionalization is the process by which a gene acquires a new function after a gene duplication event.
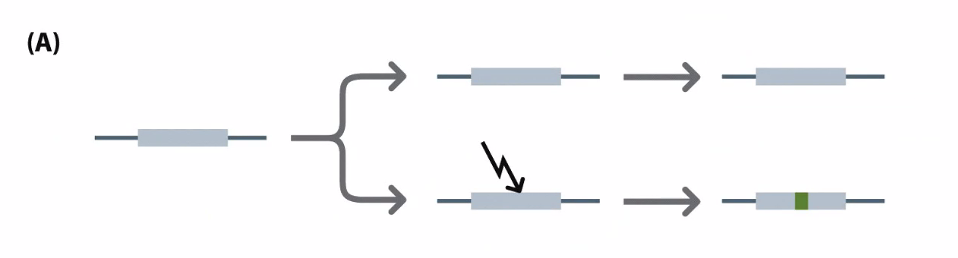
Subfunctionalization is a neutral mutation process in which each paralog retains a subset of its original ancestral function
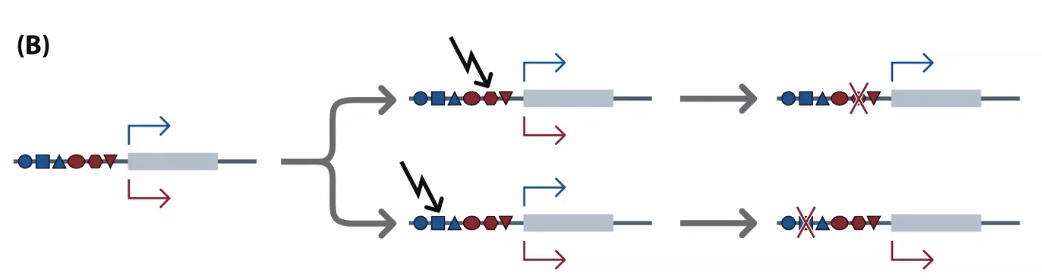
The part upstream of the gene
Pseudogenes: genes that have been copied and are now not functional
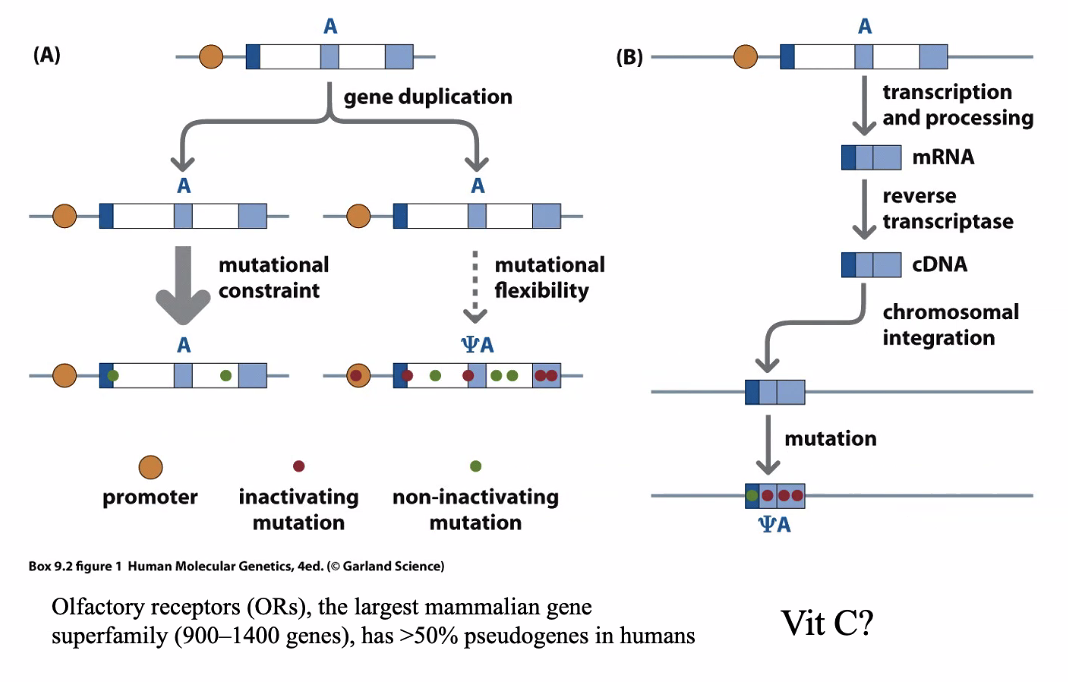
Mutation in the stop codon or other critical region
In B, these are generated from reverse transcribed mRNA
Processed pseudogenes
You can tell if it’s processed by whether it has exons — if it’s processed, it will have already been spliced
Gene to make our own vitamin C is a pseudogene
Limited to primates and bats
This is why we need dietary vitamin C
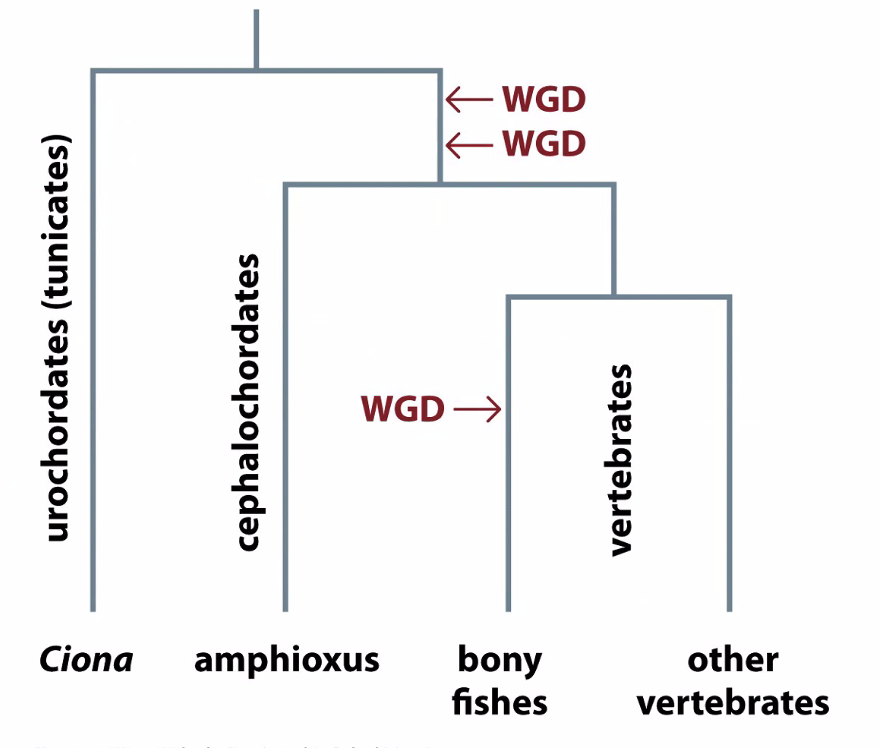
Whole genome duplications
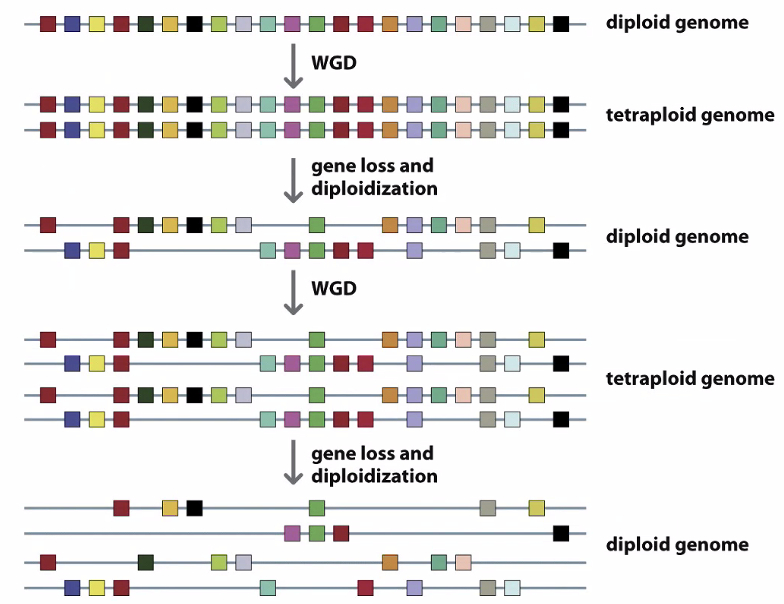
How do we know these are whole genome duplications and not individual gene duplications
Genes that are similar to each other interact — form gene families
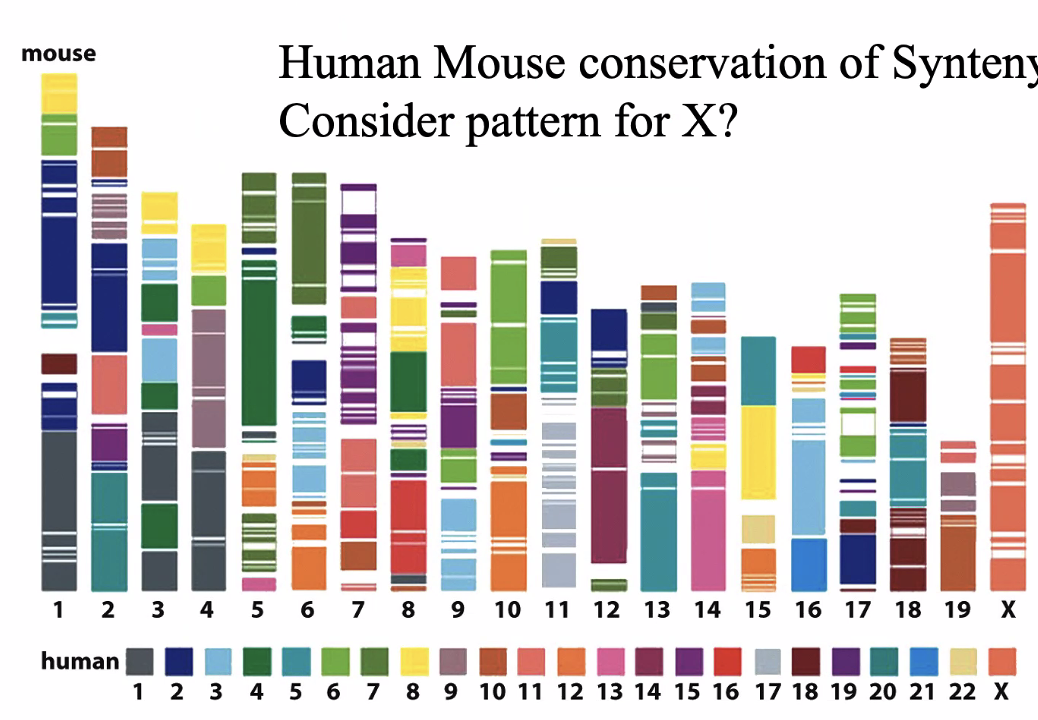
X chromosome is completely conserved
Due to Barr bodies — silencing one x chromosomes
X genes on mice are probably in X genes in humans
Functional diversity of DNA in humans
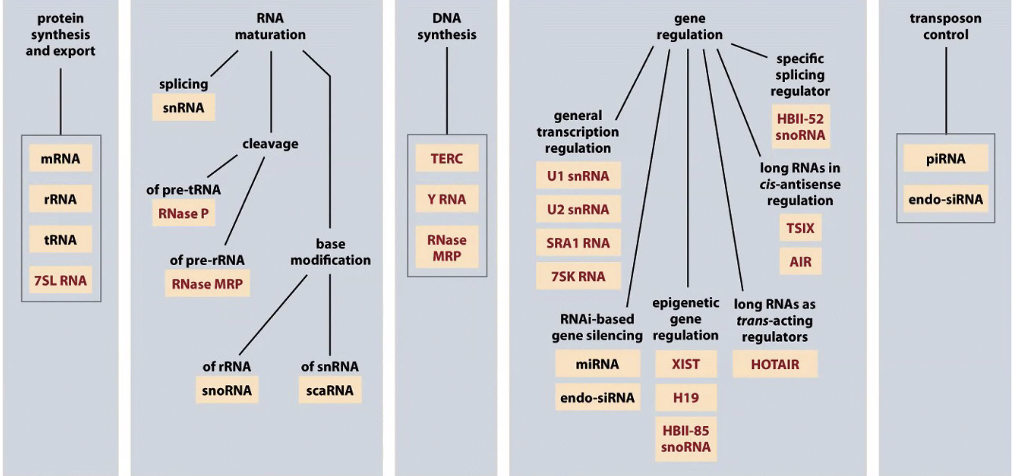
There are RNAs that control gene function and regulating transcription/translation
There are probably 2x as many genes coding for RNAs as proteins
genome organization
Transposons
Transposon repeats make up 40% of your genome

There are a lot of diseases associated with LINE-1 family elements
Alu family was one of the biggest barriers to sequencing the genome
Why do we have so many? A few ideas:
these are parasites — they care about their own replication and are successful because they can move around the genome and duplicate
DNA methylation evolved to silence these transposons
These are advantageous because they are a source of variation
Model organisms
Used to understand facts of biology and evaluate potential therapies
Comparing genome sequences provides genome-wide information on relatedness of DNA and protein sequences
Used to infer function by orthology
harder with more evolutionary difference
What makes a good model?
Easy to grow
Easy to manipulate
Not sympathetic
Closely related to humans
Assigning function to human genes
We cant experiment on humans
Model organisms are great for experiment advancing our understanding how a protein functions
We can identify the same protein in human genomes and assign the function determined in the model
50% of human gene functions come from yeast
Dogs are good for understanding genes that code for morphological and behavioral differences between breeds
Chondrodysplasia: a collection of diseases that can affect a person's stature
Studied in dogs
All small breeds have FGF4 retrogene on CFA12
Genes across species
Odine’s curse: congenital central hypoventilation syndrome
Affects the autonomic nervous system
Hypoventilation — children with this disease need a thrach to survive
Hirschprung disease
Caused by a mutation in the PHOX2B gene (4p12), only gene associated with it
Autosomal dominant
Homeobox gene family
Made up of 235 genes and 65 pseudogenes
transcription factors
Homeodomain: DNA binding region (helix-turn-helix)
Expression of these genes during development follows same patterns in model organisms
Can also lead to mixed up limbs and in model organisms
In people, they can impact development of digits
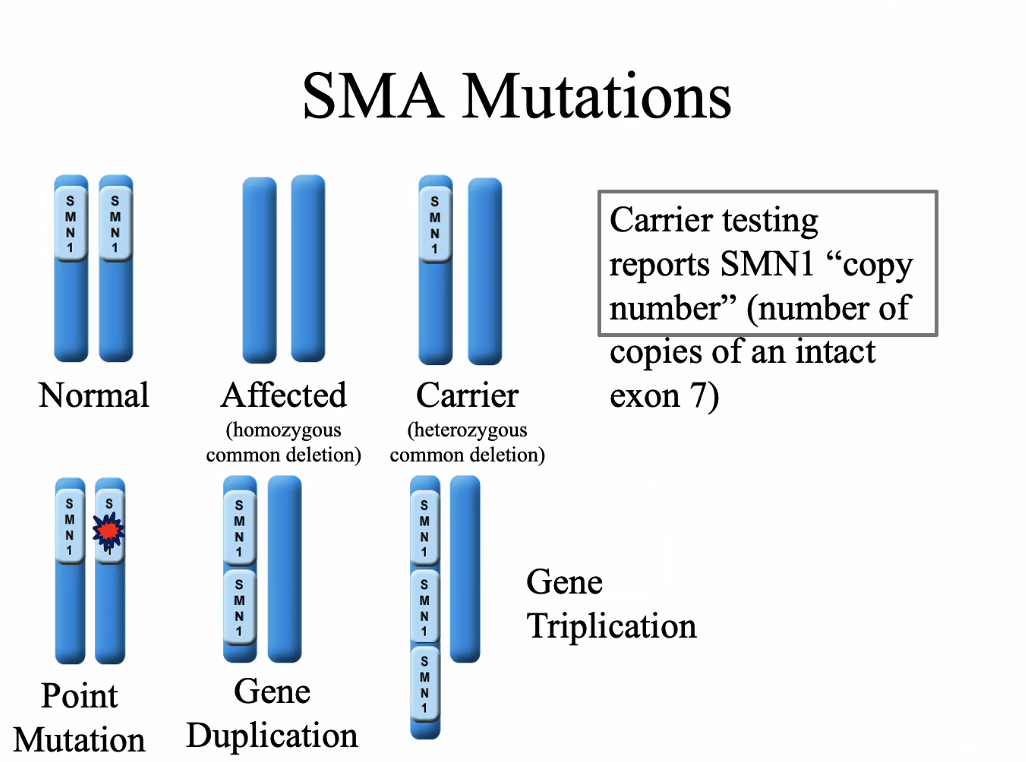
Extra SMNs — case study
SMA
SMN1 gene, 5q13.2
95-98% are homozygous for a deletion or truncation of exon 7
62% of SMN1 genes are composed of repetitive elements
Autosomal recessive
Degeneration of motor neurons
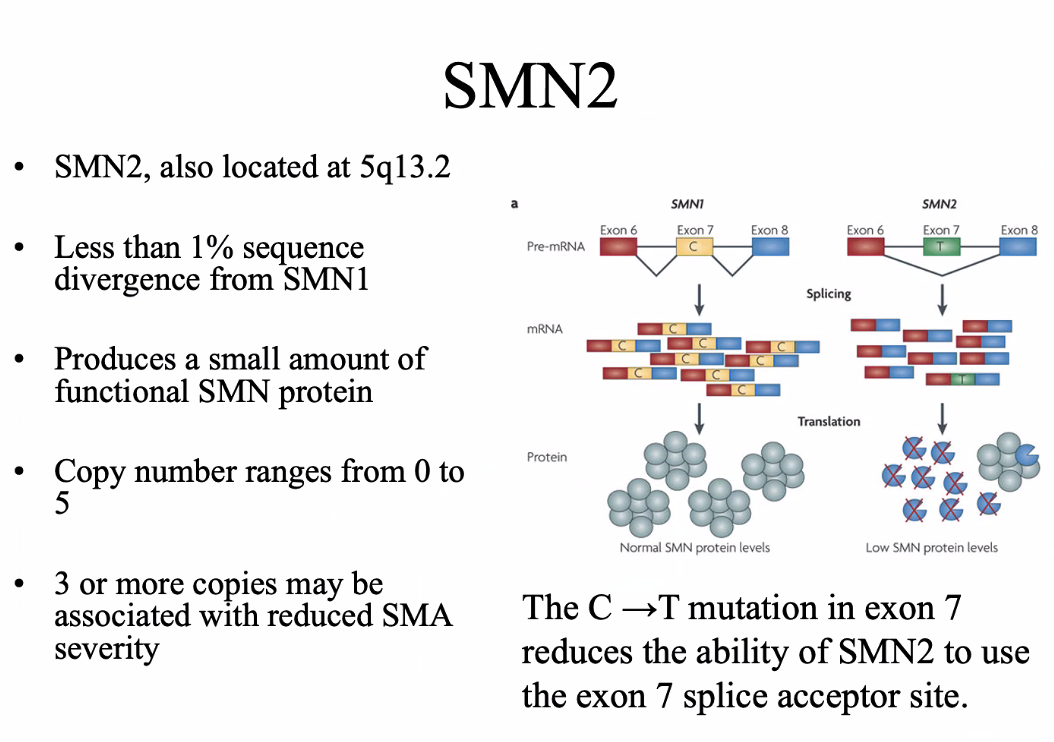
Evolution of the SMN gene
Mice and rats only have one SMN gene → the inverted duplication arose after humans diverged from mice

Sickle cell anemia
20A→ T mutation HBB gene — 11p15.4
Autosomal recessive
Sickled cells clog small blood vessels
important in malaria resistance
betal globin gene family

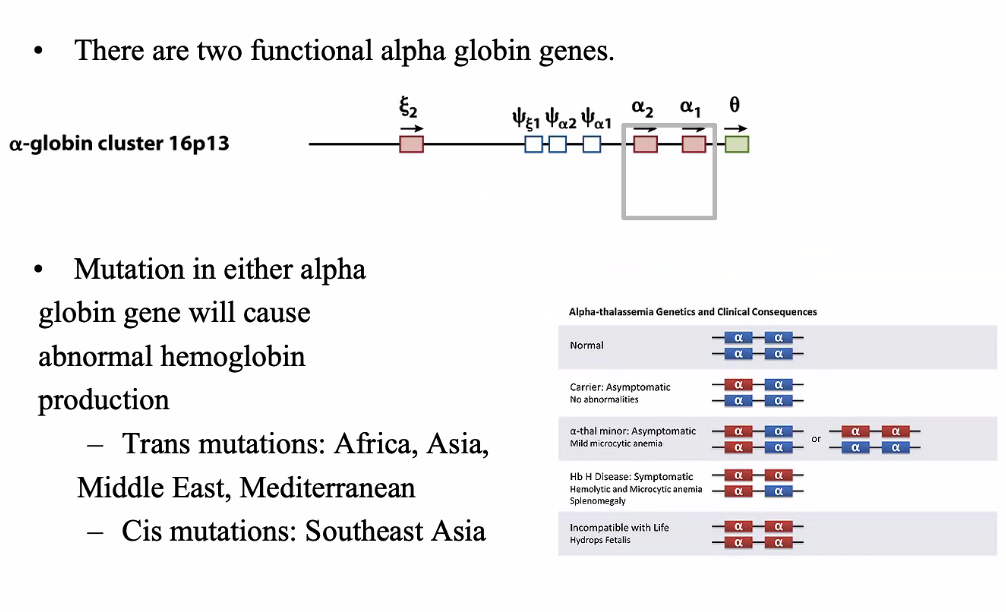
Alpha globin gene family
Apert syndrome
Apert syndrome is caused by mutations in FGFR2 gene
Craniosyntosis, midface hypoplasia, syndactyly of hands and feet, fusion of cervical vertebrae
There are 7 other syndromes caused by the same mutations
Most mutations are missence mutations
FGFR2 shows alternative splicing. A known Alu insert in exon IIIc inappropriately promotes splicing to form FGFR2
Lil bub
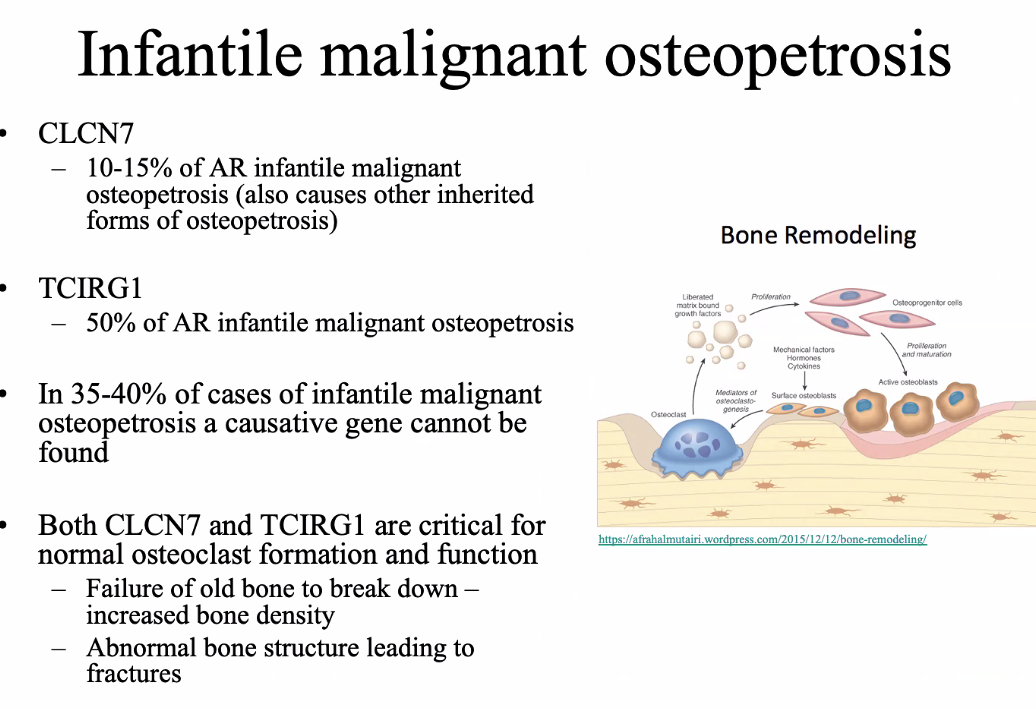
homozygous deletion of exon 8 of the TNFRSF11A gene
Encodes RANK protein
3/13/2025
Types of variation
SNPs: Changes to a single nucleotide within the DNA
sequence, such as single base substitutions, insertions, or deletion
Deletions + duplications: Missing or extra genomic information. Both very small (single nucleotides) and very large (whole chromosomes)
Indels
CNVs
Trinucleotide repeat expansion: increased number of trinucleotide repeats (three specific nucleotides) in a given area of a gene (e.g., Friedreich ataxia, Huntington’s)
Methylation: The addition of methyl groups to DNA can turn the activity of a gene off. Changes to the methylation pattern can cause disease (e.g., imprinting disorders such as Angelman syndrome). These changes do not change the DNA sequence.
Mitochondrial DNA variants: Changes to the DNA in the mitochondria can include single nucleotide variants and deletion/duplications (e.g., mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes or MELAS).
Nomenclature
There is a standard way to communicate human genetic variation necessary to make use of that data
Each variant has a unique identifier (e.g. rs212570 rs = “reference SNP”)
These variants can be described using a set of common conventions. (e.g. g.1162G>A or p.F508del).
g-genomic, c-cDNA, r-rRNA, p-protein
Molecular mechanism of pathology
Loss-of-function mutations: gene products have no or reduced function.
Can display haplo-insufficiency
Allelic homogeneity (all mutations with same phenotype)
Gain-of-function mutations: gene product does something positively abnormal.
Often due to abnormal product interactions, increased expression or expression in the wrong place.
Pathogenic: Variant has been shown to disrupt gene function in the lab, is strongly associated with disease in humans, and does not appear in the general population
Likely pathogenic: Variant is predicted to disrupt gene function is seen in some individuals with disease, and does not appear in the general population
Variant of uncertain significant (VOUS): Evidence for variant pathogenicity is conflicting or absent
Likely benign: Variant is not predicted to disrupt gene function and has not been seen in individuals with disease.
Benign: Variant has been shown not to disrupt gene function and is present in high frequency in the general population
Funtional consequences of a variant
Missense mutation
Frameshifts
Nonsense mutations
Mutations that affect splicing
Mutations that affect expression
Short tandem repeats
Central dogma
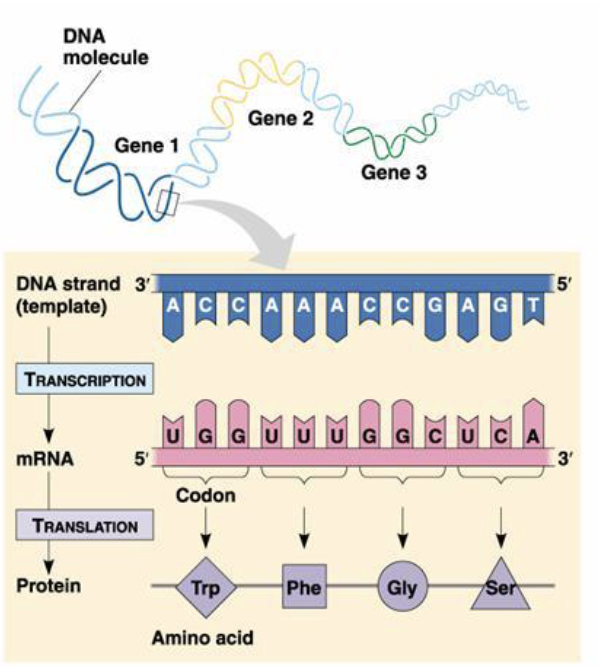
Missense mutation
SNP that leads to change in the protein
Nonsense mutation
Mutation that gives rise to a stop codon
Dynamic mutations
SHort tandem-repeat polymorphic loci become unstable above threshold size
Many but not all are pathogenic
Can display anticipation increased severity or lower age of onset in successive generations
Case study
It is 1968 and George and Lorraine asked you to
evaluate their two oldest children, Dave and Linda.
They report that Dave has always been a “sickly” child
and he was not able to attend kindergarten due to his
frequent respiratory infections. Linda is also often ill
but her parents are mostly concerned about her size; she
is less than the 5th percentile for height and weight.
George and Lorraine also have a 4 month old son,
Marty. Linda tells you that Marty is a healthy boy but
she thinks it is very odd that she always tastes salt when
she kisses him. George is of Irish descent and Linda is
of French and English descent



Kabuki syndrome
KMT2D (12q13.12) and KDM6A (Xp11.2) genes
Characteristic facial features
Arched eyebrows
Long eyelashes
Long palpebral fissures
Flat tip of nose
Large earlobes
Intellectual disability
Siezures
Microencephaly
Hypotonia
Autosomal dominant inheritance
Finding the gene
Whole-exome sequencing of 10 individuals with Kabuki syndrome (unrelated) with varied ethnic backgrounds
Why was it so hard to find the gene?
Locus heterogeneity
Failure of WES to find the mutations in the targeted exome
Causative mutations are outside the targeted exome
How was this addressed?
Use less strict criteria
Found 16 varients shared in at least 7 patients
MLL2 (KMT2D) — 7 cases had mutations
Loss of function found in 9
What is MLL2
FOund in drosophila nad mouse
Involved with histones and chromatin regulation
MLL2 provides instructions for making an enzyme called lysine-specific methyltransferase 2D
Loss of MLL2 is an embryonic lethal in mice
What does finding genes do for patients?
Allows for earlier diagnosis and treatment
Prenatal testing
Confirmation of diagnosis
BEtter understanding of molecular mechanisms
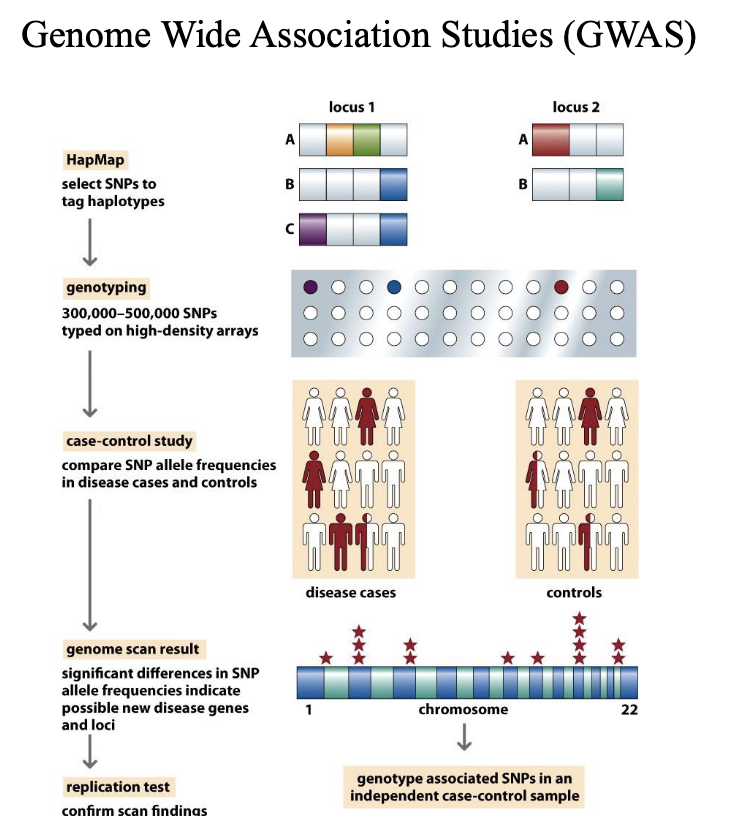
Genome-wide association studies (GWAS)
Identify genetic variants (Haplotypes) associated with a trait or illness.
Mainly use SNP Chip technologies like 23 and me rather than whole genome sequencing.
GWAS typically involve large numbers of individuals – GIANT consortium use 5.4 million human data sets
Haplotype
A set of DNA variations, or polymorphisms, that tend to be inherited
together. A haplotype can refer to a combination of alleles or to a set of single nucleotide polymorphisms (SNPs) found on the same chromosome.
They are transmitted throughout the pedigree as a block without recombination.
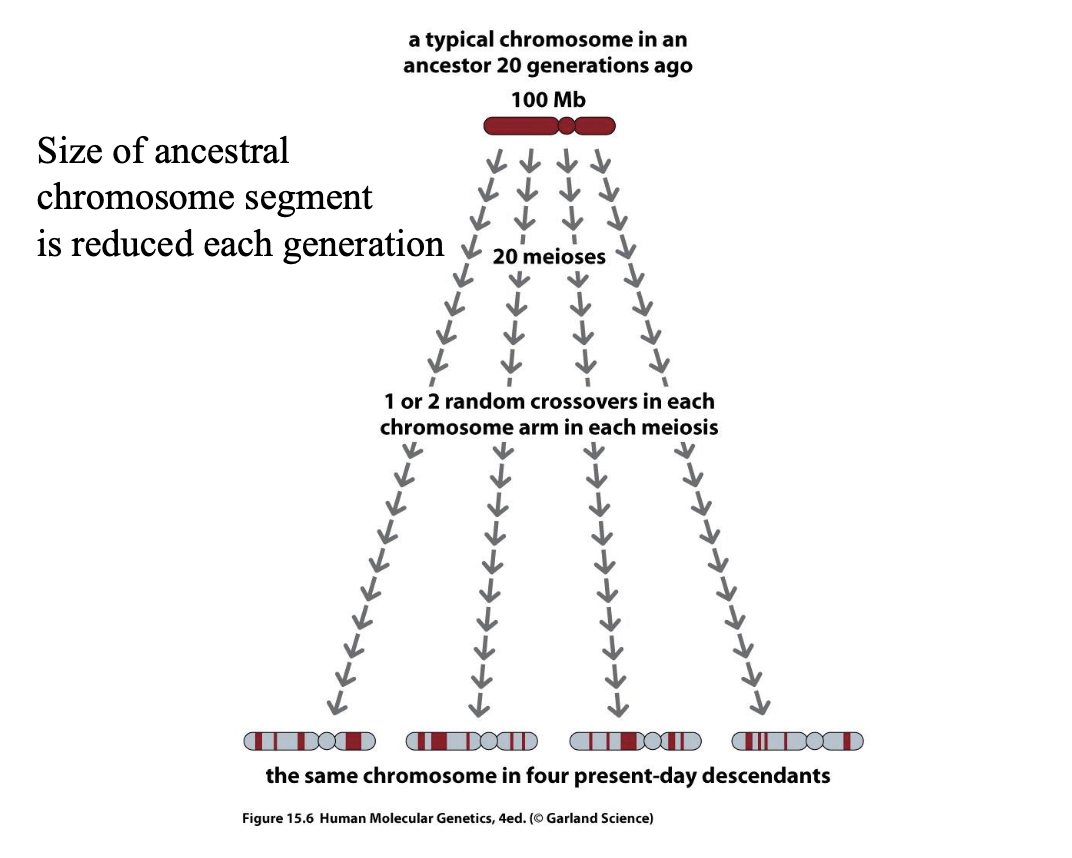
What determines the size of a haplotype?
– Age of the segment
– Local rate and pattern of recombination
Mapping genes conferring susceptibility to complex diseases
Major genetic effects on human health are complex (many
genes).
An approach to finding susceptibility factors (GWAS) finds statistically significant associations between ancient haplotypes and disease.
Results are somewhat disappointing
– Common disease-common variant hypothesis
Lots of variants with small effects
– Mutation selection hypothesis
Variants with significant effect lost be selection
Problems with GWAS
Variants must be old and able to persist for many
generations to be associated with a haplotype shared by
many.
Variants with strong effects are removed from the
population by selection but replaced by new mutations
in different haplotypes.
May be different strong mutations in genes from same
pathway making them untraceable by linkage because
genes for similar process are not clustered
Variants with small effects require lots of cases to find
statistical significance.
Four basic gene therapy approaches
Gene replacement: the delivery of a functional gene to replace a non-working gene
Gene silencing: inactivation of a mutated gene that has become toxic to cells
Gene addition: over expression of a “foreign” or exogenous gene to impact cellular function
Gene editing: a permanent manipulation of a gene in a patient’s genome.
SEE SLIDES ——- 23 and ME
CRISPR
Stands for clustered regularly interspaced short palindromic repeats
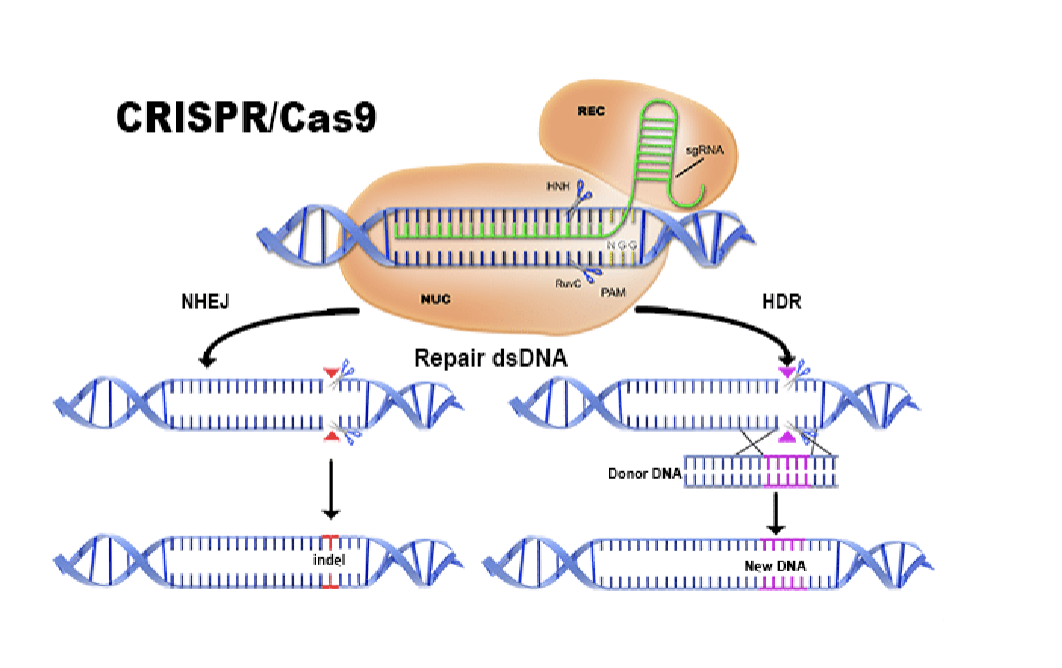
CRISPR will go to any gene that you want
Can knock the gene out by making it make errors (NHEJ)
Can give it a new DNA sequence to repair it (HDR)
Bacterial immune system
In vivo
injecting gene therapy into individual and target it
Ex vivo
Culture the cells, do gene therapy on the cells, and infuse the cells back in the patient
Gene delivery vehicles
For CRISPR: usually use viruses
Adenovial
Retroviral
Lentiviral
Adeno-assocaited viral
Viral vector components
The protein capsid defines the vector’s tissue or cell tropism and antigen
recognition
The transgene of interest, which when expressed in cells, serves to confer a desired effect
The“regulatory cassette” that controls expression of the transgene as an episome or as a chromosomal integrate
Dont need to memorize below



3/25/2025 — pharmacogenomics
HIstory/evolution of adaptations to xenobiotics
Substances that are foreign to the body or an ecological system
The great oxygenation event (GOE) 2.45 BYA nearly wiped out obligate anaerobes probably gave rise to the enzyme families like P450
Genetic solution (adaptation) to xenobiotic toxin (oxygen)
The great Animal Plant Wars last 800 Million years
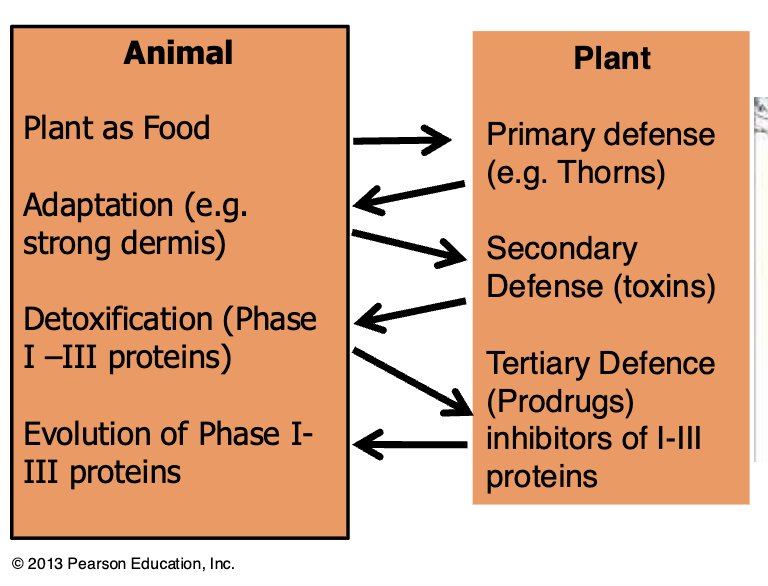
Example: possums vs eucalyptus
Eucalyptus is a potential food source for possums and koalas
Eucalyptus expresses terpines (toxic)
Possums feeding on Eucalyptus express >50 more CYP450 increasing metabolism and clearance of terpines by order of magnitude
Evolution of drug metabolism as a science
Morphine was discovered by Freidrich Wilhelm Adam Serturner
(1783-1841), a 21-year-old pharmacist’s assistant. He wondered
about the medicinal properties of opium, which was widely used by
18th-century physicians
Began transformation of pharmaceutical chemistry from alchemistry
Richard Tecwyn Williams
1942, worked on the metabolism on TNT with regard to toxicity
in munitions workers; due to the war he assembled teams to work on metabolism of sulfonamides, benzene, aniline, acetanilide, phenacetin, and stilbesterol. Established the role of metabolism in drug action
How do drugs work
Pro drug: compound that you take that you have to metabolize for it to be active. The metabolites from your system produce the bioactive compounds
Active drug: The drug you are taking is in the active form, but it has to get where you want it to go before it metabolizes
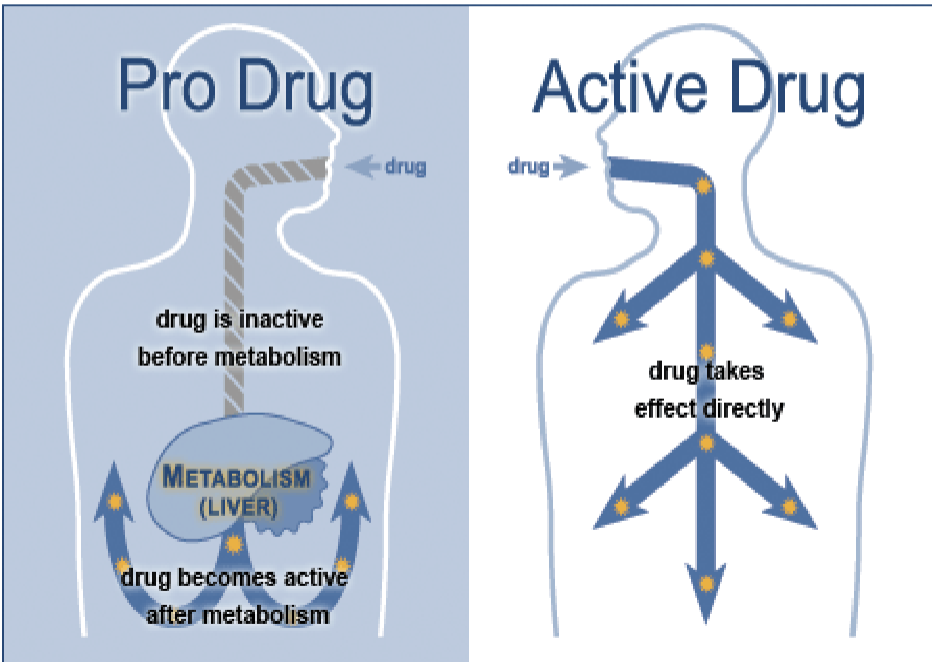
Biotransformation of drugs into active metabolites

Organs of drug metabolism
Liver — most common
Kidneys
Muscle tissue
Intestinal wall
Lungs
Skin
Blood
Main ways of biotransformation of drugs
Dont need to memorize

We have a whole bunch of genes that make proteins (enzymes) that metabolize drugs
Pharmacogenomics
Pharmacogenomics: study of how an individuals entire genetic makeup determines the body’s response to drugs
Core concept: because so many interactions occur between a drug and proteins within the patient, many genes and many different genetic polymorphisms can affect a person’s response to a drug
Knowing someone’s genotype should help inform drug therapies
Optimizing drug therapies
On average, a drug will be effective in only about 50% of patients who take it
Trial and error is used until the correct drug is found for a patient
This is a waste of time and can be dangerous
Pharmacogenomics increases the efficacy of drugs by focusing drugs on subpopulations of patients who will benefit
Personalized pharmacogenomics is already widely practiced in diagnosis and treatment of cancer
It is important to understand each patient’s genetic profile to select an appropriate treatment — particularly those newer treatments based on molecular characteristics of tumors
HER-2
Personalized medicine was successfully used in the HER-2 gene and the use of the drug Herceptin in breast cancer
The human epidermal growth factor receptor 2 gene is located on chromosome 17 and codes for transmembrane tyrosine kinase receptor protein called HER-2
In about 25 percent of invasive cancers, the HER-2 gene is amplified and the protein is overexpressed on the cell surface.
In some breast cancers, the HER-2 gene may have as many as 100 copies per cell.
This amplification is associated with increased tumor invasiveness, metastasis, and cell proliferation as well as poorer patient prognosis
HER-2 is an oncogene
THerapy
Using recombinant DNA technology, Genentech Corporation in California developed a monoclonal antibody known as trastuzumab (or Herceptin®).
When bound to the receptor, Herceptin appears to inhibit the signaling capability of HER-2 and may also flag the HER-2 expressing cell for destruction by the patient’s immune system.
In cancer cells that overexpress HER-2, Herceptin treatment causes cell-cycle arrests, and in some cases, death of cancer cells.
Optimizing therapy
Herceptin only acts on breast cancer cells that have
amplified HER-2 genes; therefore it is important to
know the HER-2 phenotype of each cancer.
Herceptin has potentially serious side-effects;
therefore its use must be limited to those who could
benefit from the treatment.
The treatment is directed toward “HER2 Positive”
tumors.
Reducing adverse drug reactions
Every year, about 2 million people in the US experience serious side-effects from pharmaceutical drugs
The costs associated with these adverse drug reactions (ADRs) are estimated to be $136 billion annually.
Sequencing variations ina large number of genes can affect drug responsiveness
Cytochrime P450 families of enzymes are particularly significant and are encoded by 57 genes
Superfamily of heme enzymes. Can catalyze different reaction types, mainly hydroxylation
Human: 18 families, 43 subfamilies, 57 sequenced genes
can be induced and inhibited
Occur in most tissues (except muscles and blood)
Exhibit genetic polymorphism (atypical biotransformation
Nomenclature:
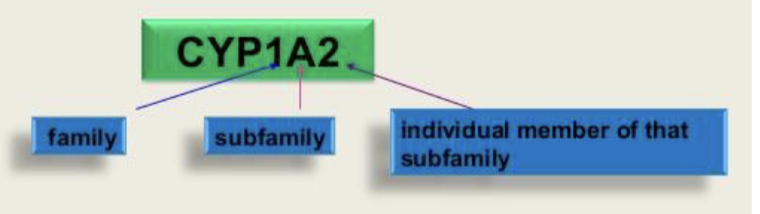
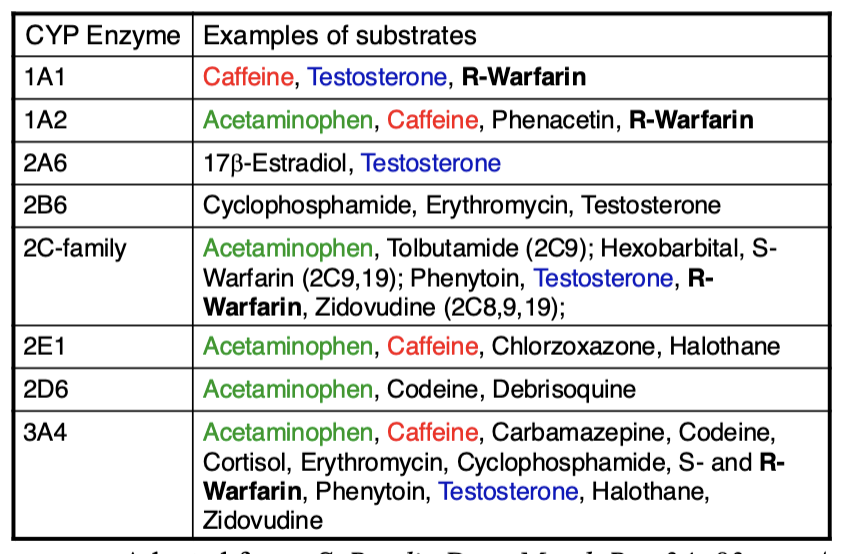
Important human liver drug CYP450s
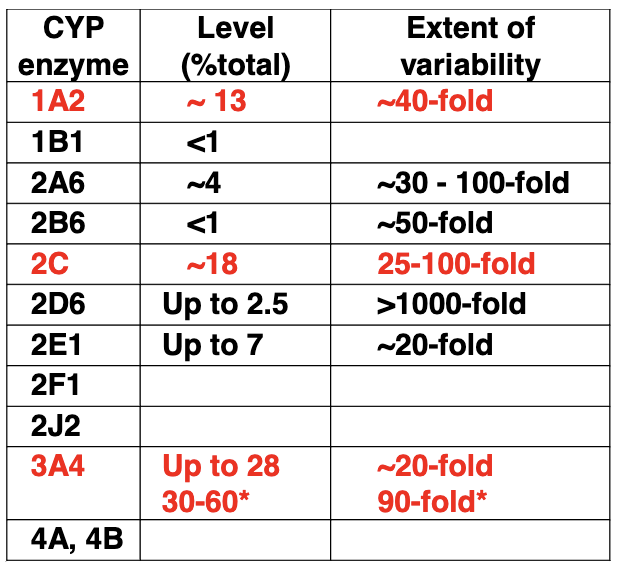
Drugs and grapefruit juice
Grapefruit juice interacts with CYP450s
Can block enzyme that degrades drug in drugs that are active when you take it — can lead to overdose
Can block transporter that brings drug to body’s cells, leads to too little drug
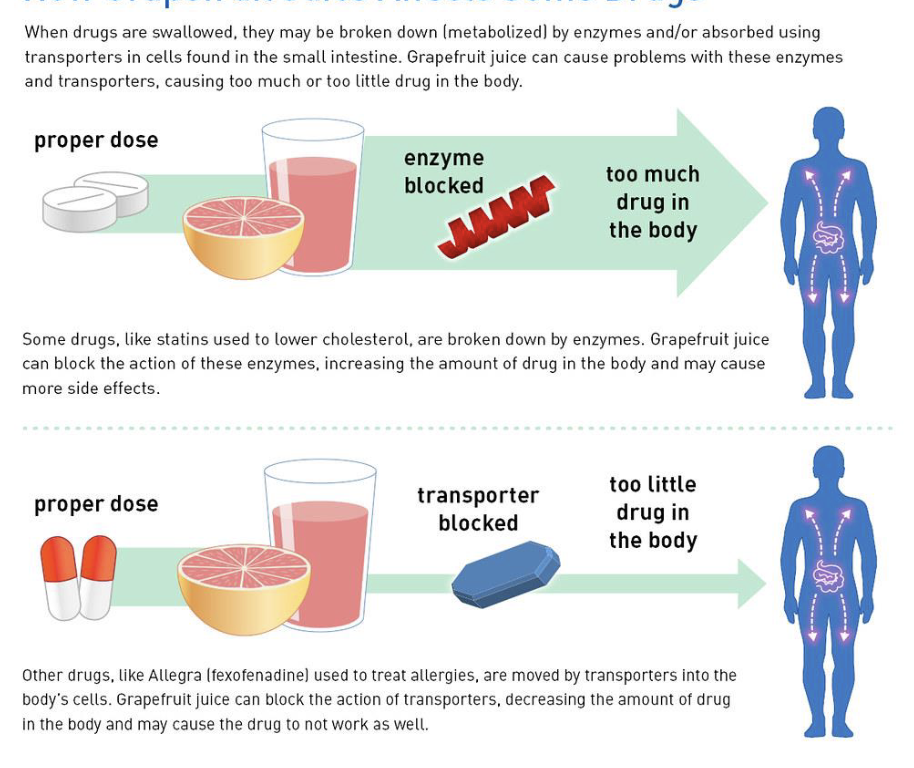
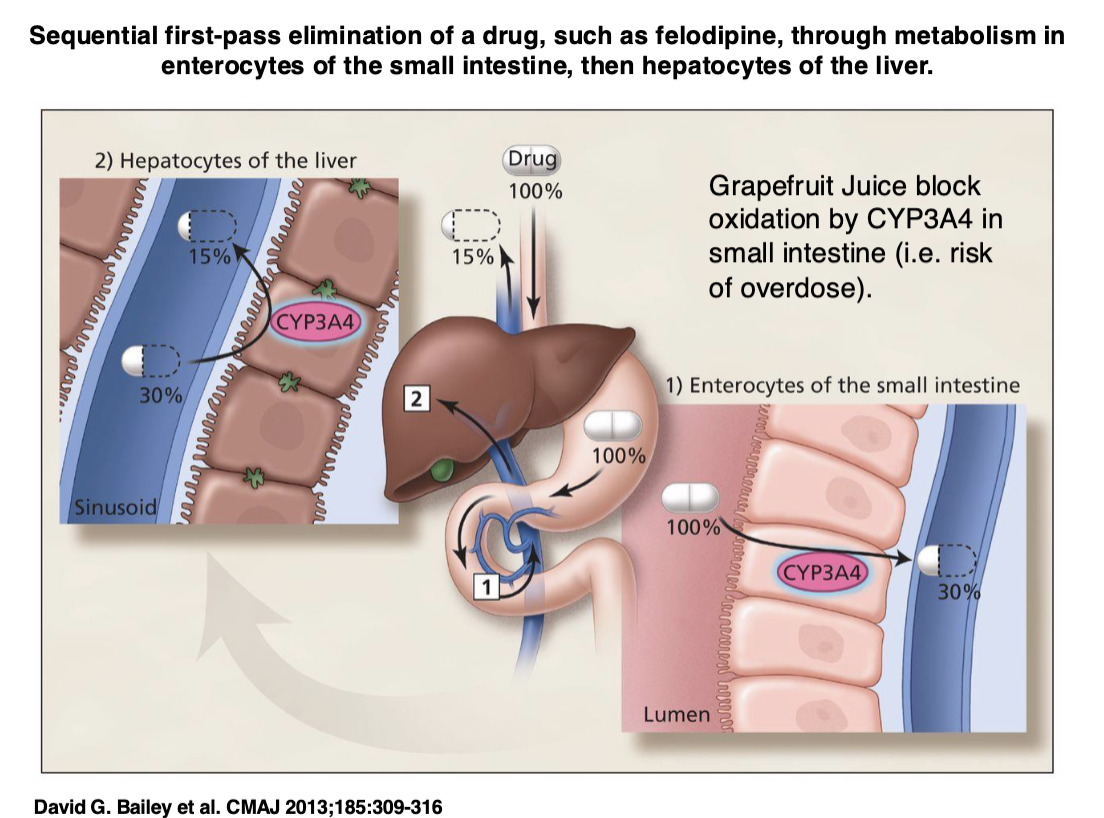
LEFT OFF HERE IN FLASHCARDS
Reducing adverse drug reatctions
People with some cytochrome P450 gene variants metabolize and eliminate drugs slowly, which can lead to accumulations of the drug and overdose
In contrast, other people have variants that cause
drugs to be eliminated quickly, leading to reduced
effectiveness.
Example:
There are more than 70 variant alleles of the gene CYP2D6 which encodes the debrisoquine hydroxylase enzyme. This enzyme is involved in the metabolism of approximately 25 percent of all pharmaceutical drugs, including diazepam, acetaminophen, clozapine, beta blockers, tamoxifen, and codeine
Another example of pharmacogenomics in personalized medicine is that of the CYP2C9 and VKORC1 genes and the drug warfarin.
Warfarin (Coumadin) is an anticoagulant drug prescribed to prevent blood clots after surgery and to aid people with cardiovascular disease who are prone to blood clots.
Warfarin inhibits vitamin-K-dependent synthesis of several clotting factors.
If the dosage of warfarin is too high, the patient may experience serious hemorrhaging; if it is too low, the patient may develop life-threatening blood clots.
20 percent of patients are hospitalized during
their first six months of treatment due to warfarin side-effects.
Two single-nucleotide mutations in CYP2C9 lead to reduced elimination of warfarin and increased risk of hemorrhage.
Patients who are heterozygous or homozygous for some alleles of CYP2C9 require a 10–90 percent lower dose of warfarin.
The FDA has recommended the use of CYP2C9 and VKORC1 genetic tests to predict the likelihood that a patient may have adverse effects to warfarin.
It is estimated that the use of warfarin genetic tests could prevent 17,000 strokes and 85,000 serious hemorrhages per year.
Acetaminophen pathway
APA is an active drug. SOme metabolites are hepatotoxic
Acetaminophen belongs to a class of drugs called analgesics (pain relievers) and antipyretics (fever reducers). The exact mechanism of action of acetaminophen may reduce the production of prostaglandins that cause inflammation and swelling.
Why do we need to understand how genes function?
Different mutations can cause the same disorder
Knowing what specific mutations a person has can direct effective treatment
This may be the most useful outcome of personal genomics
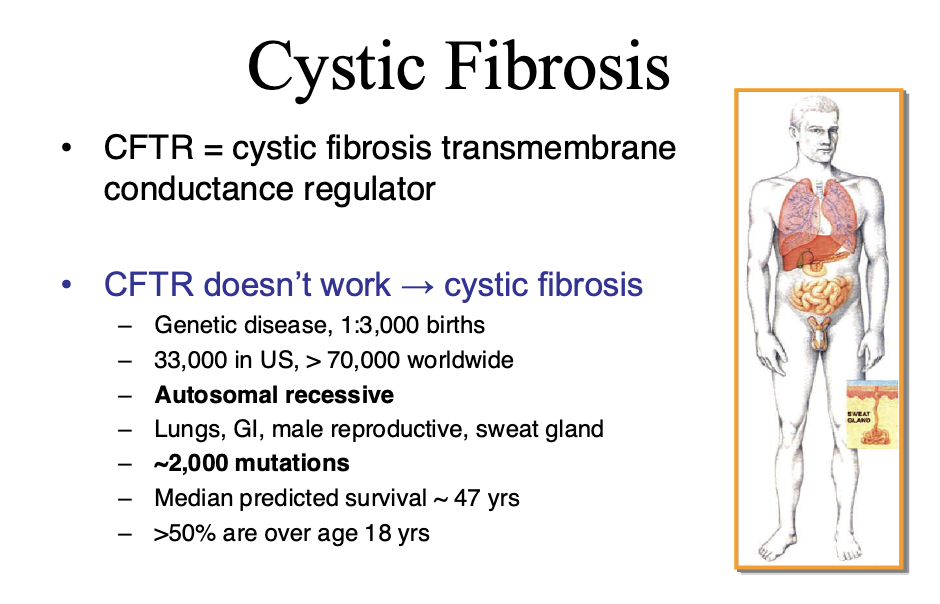
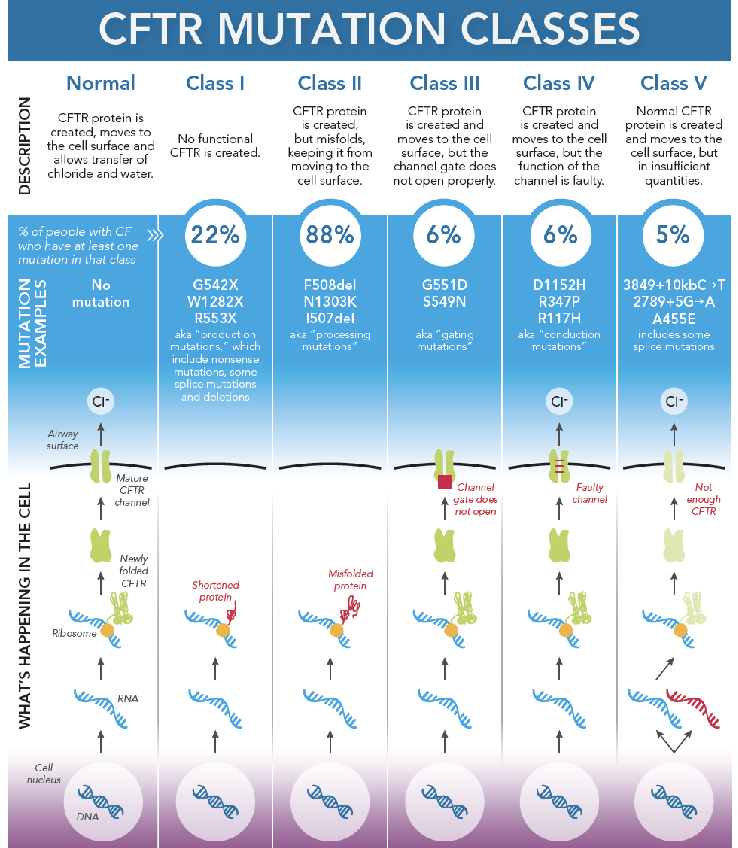
Ex cystic fibrosis is caused by thousands of genes. It is useful to know which gene it is caused by
CFTR: cystic fibrosis transmembrane
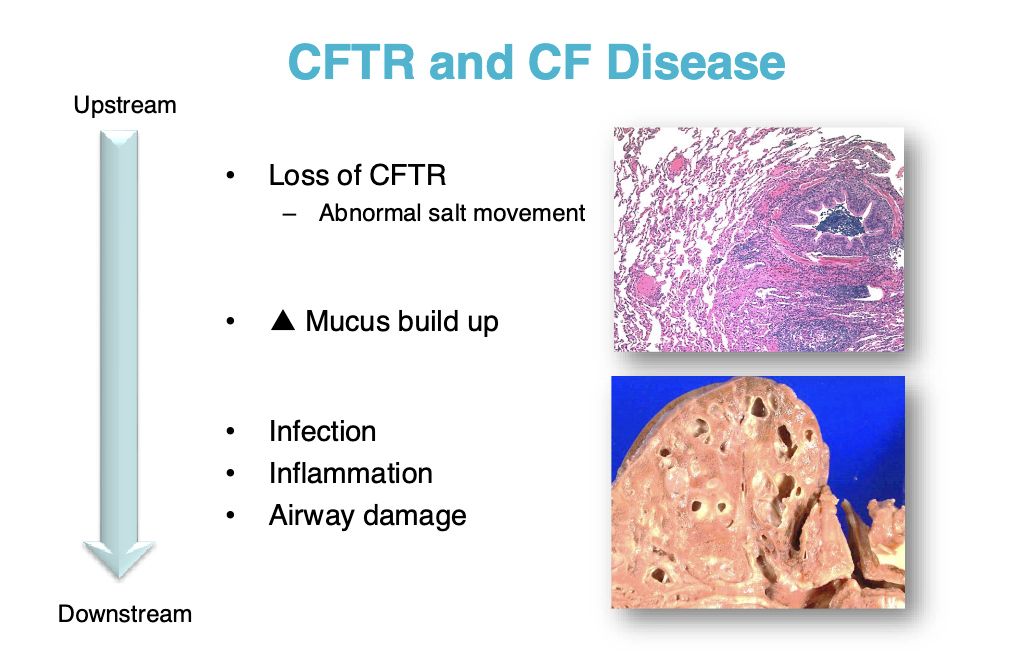
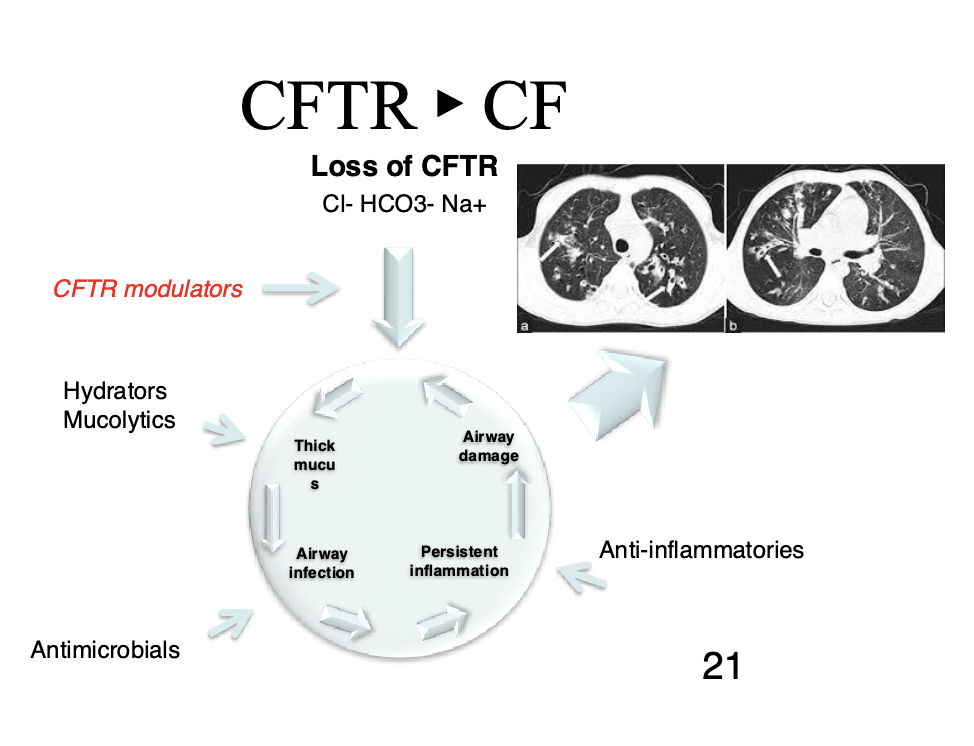
CF is caused by dysfunctional CFTR
Leads to infection, inflammation, and airway damage
We usually treat this by treating the symptoms (breaking up the mucus, antimicrobials, etc.)
We would like to be able to treat the genetic defect more directly
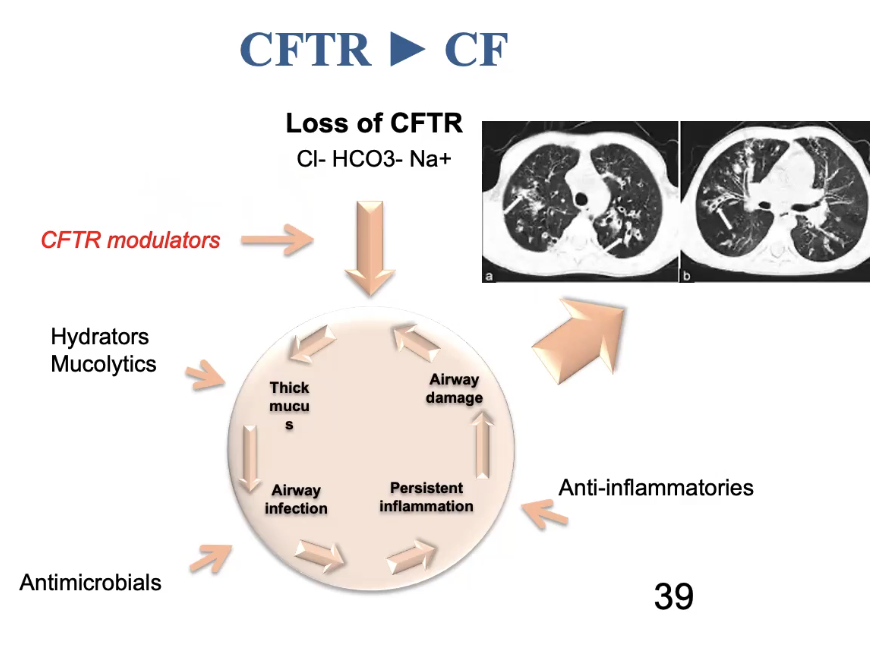
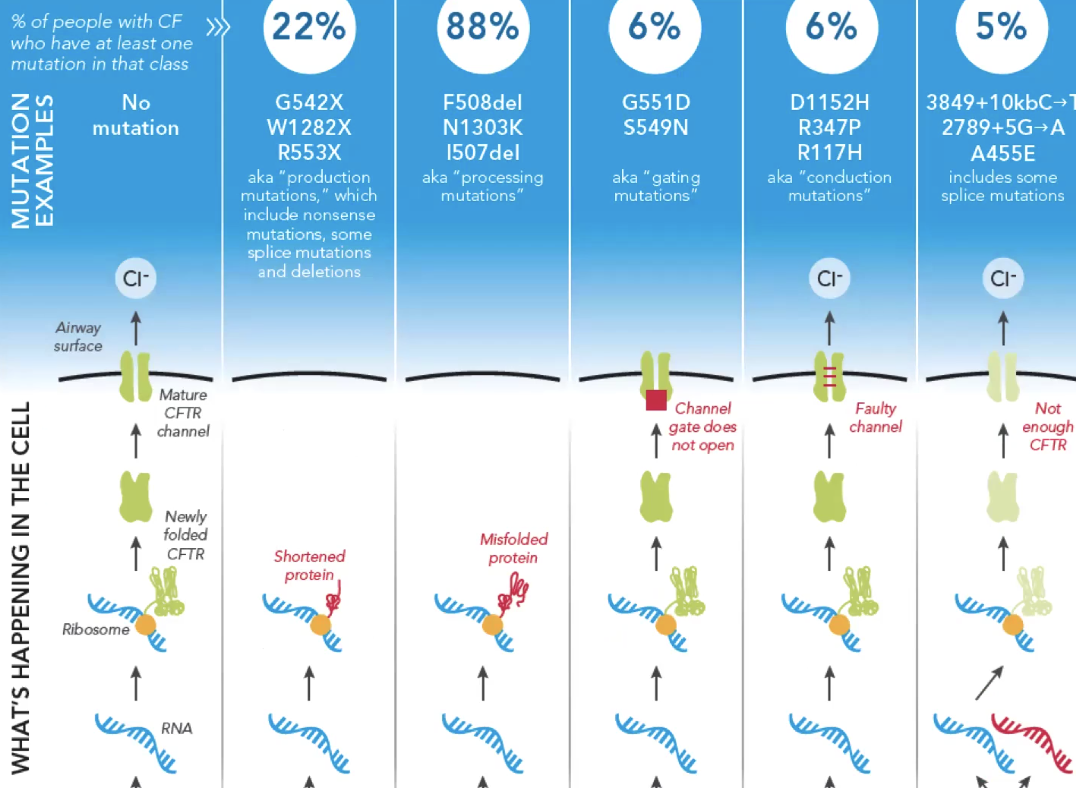
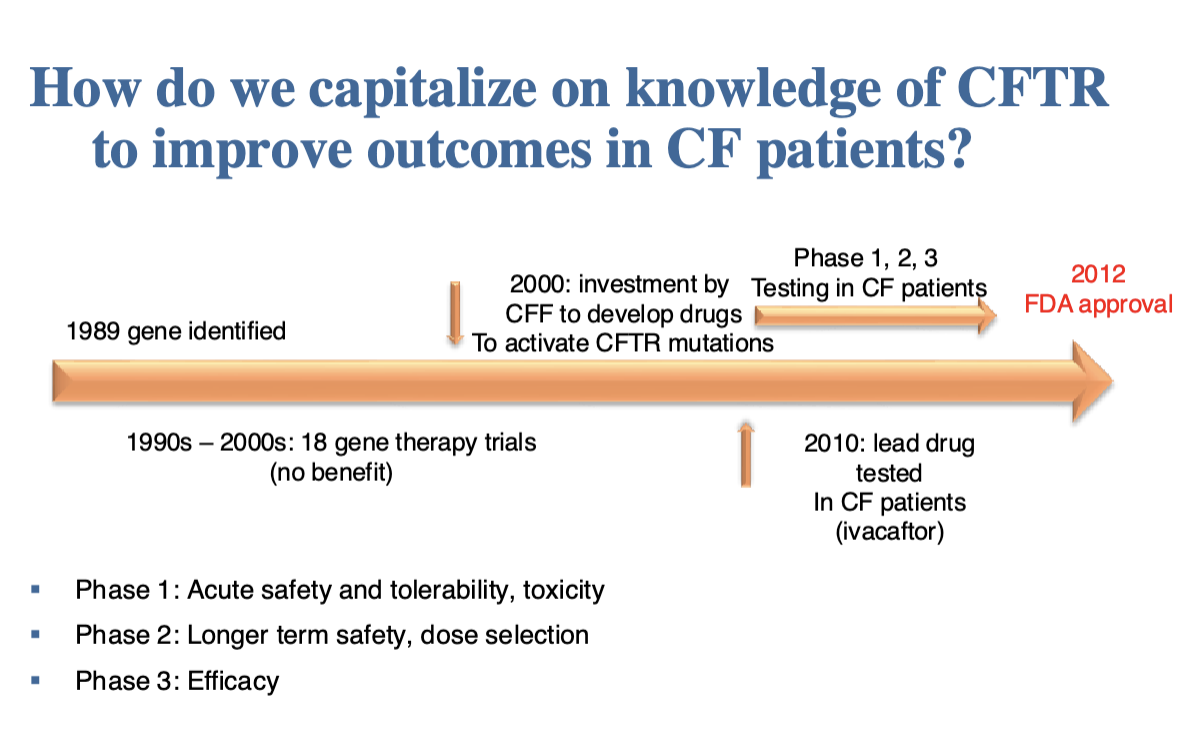
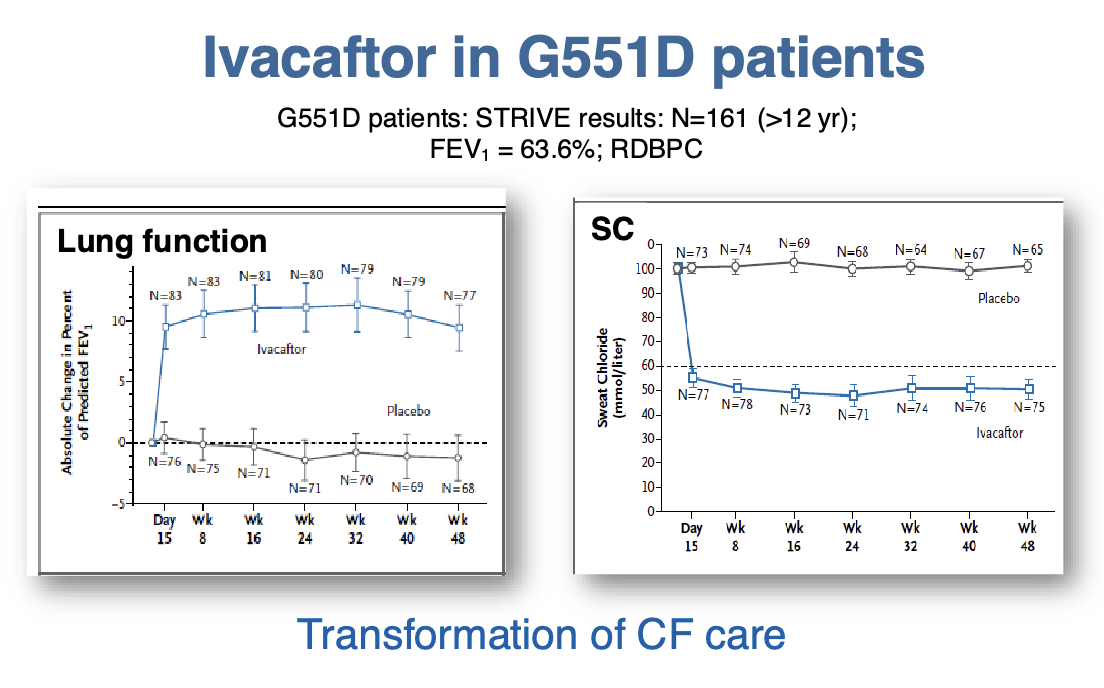
Critical concepts for the future of pharmacogenomics

Phenylalanine hydroxylase deficiency (PKU)
PAH gene, 12q23.2
Autosomal recessive
Deficiency of phenylalanine hydroxylase
Inability to process phenylalanine
Characteristics (untreated)
Severe intellectual disability
Epilepsy
Microcephaly
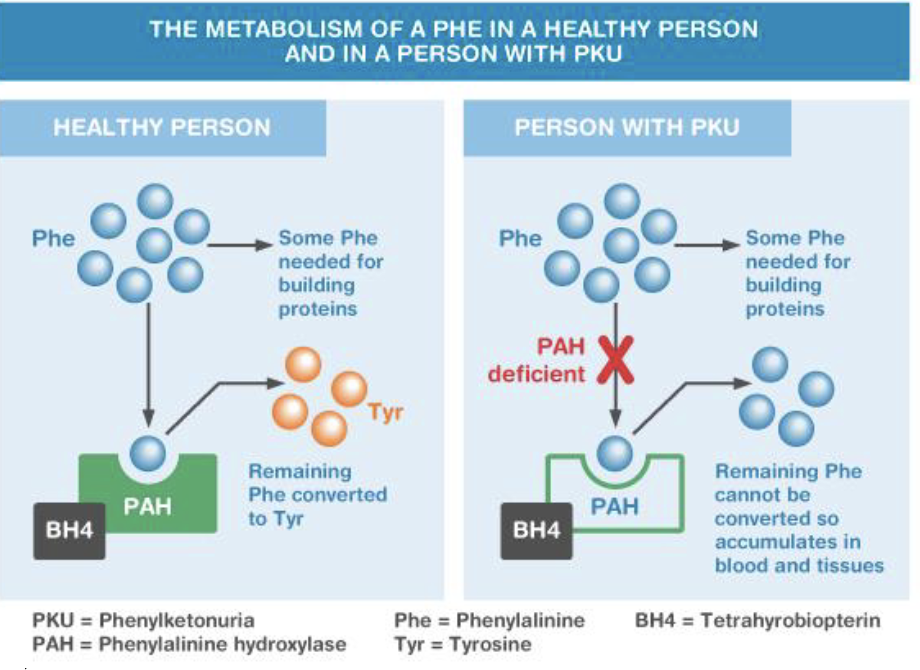
Treatment
Restriction of dietary Phe at least through adolescence
Supplementation with Phe-free formula
Can avoid negative consequences based on genetic test
Rh disease
Antigens are genetically inherited
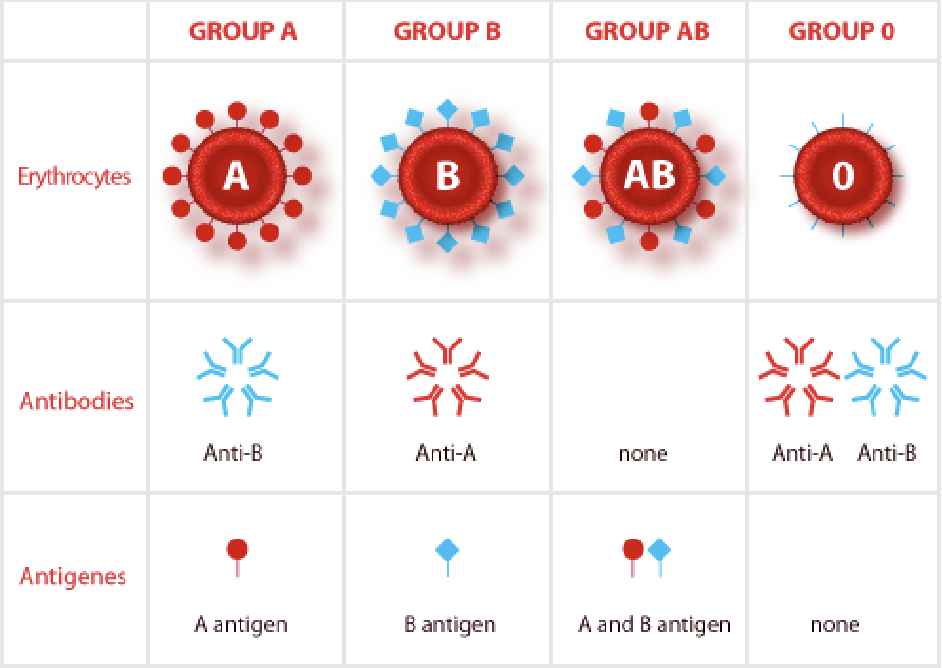
Rehus factor (Rh)
D antigen
If you have the gene for Rh, you are Rh positive (dominant)
AB+ = universal acceptor
O- = universal donor

Rh incompatibility
Transfusion of Rh positive blood to an Rh negative recipient will produce a transfusion reaction
Hemolytic disease of the newborn
Rh- mother, Rh+ father
Mother becomes sensitized to the Rh positive blood type during delivery
All subsequent pregnancies are at risk if the fetus is Rh positive — once sensitized, the mother can’t be desensitized
The immune responded will be more severe with each pregnancy
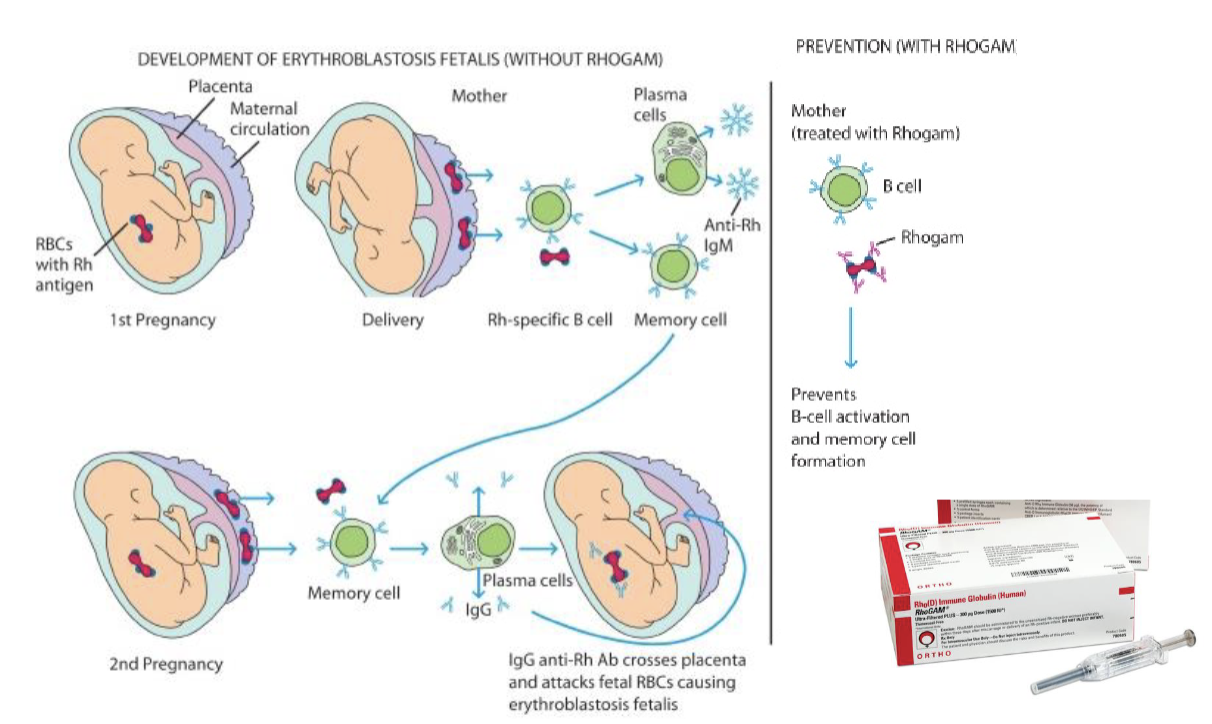
You need to know the fetus’s genotype for the treatment to be affected
Single gene disorders
Fabry disease
GLA gene, Xq22.1
Alpha-galactosidase A enzyme
Active in lysosomes, break sown Gl-3
Buildup of GL-3 is toxic to the body
X-linked
Episodes of severe extremity pain
Angiokeratomas
Eye anomalies
Heart disease
Stroke
Males more severely affected
Treatment
enzyme replacement therapy (fabrazyme)
IV infusion given every 2 weeks, may take 4-5 hours
Proven to reduce Gl-3 buildup, but limited change in overall lifespan
Most people have adverse side effects
Galafold
Not enzyme replacement
Binds to misfolded protein and refolds it
3/27/2025
What is epigenetics
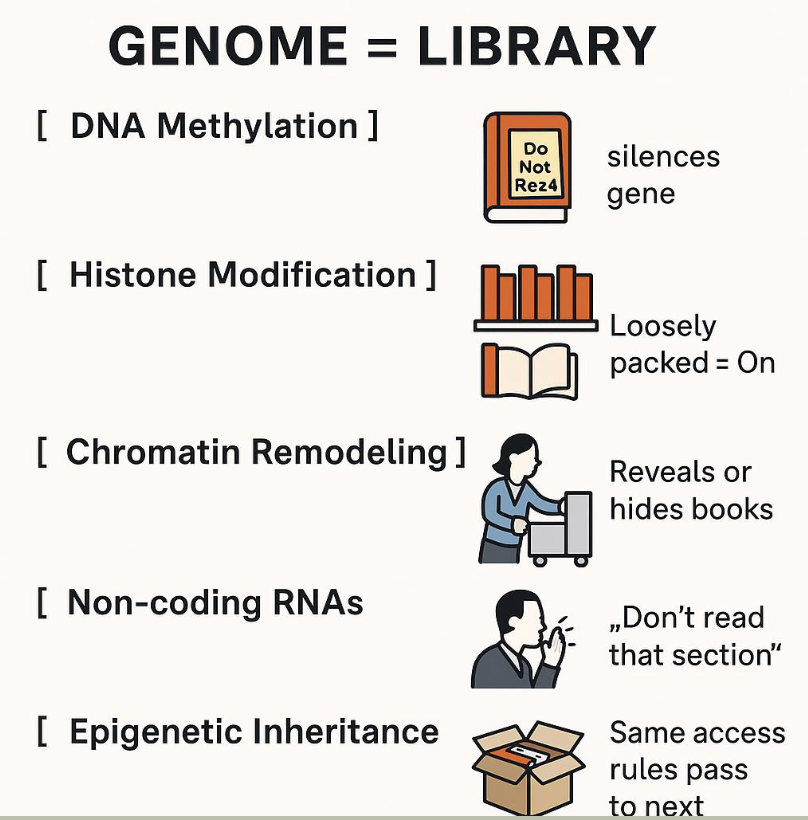
epigenetics: the study of changes in gene activity that do not involve changes to the DNA sequence itself, but still get passed on when the cells divide — and sometimes across generations. These changes affect how genes are turned on or off
Mechanisms include
Methylation
Histone modification
Non-coding RNAs
First coined by Conrad Waddington in the 1940s
Key concepts
DNA methylation
The addition of a methyl group (-CH3) to cytosine bases, usually at CpG sites
Often silences gene expression by preventing transcription factors from binding or recruiting repressive proteins
Histone Modification
Post-translational modifications (acetylation, methylation, phosphorylation) of histone proteins around which DNA is wrapped.
Function: alters chromatin structure to make DNA more or less accessible to the transcription machinery
Non-coding RNAs (ncRNAs)
Small and long RNAs that don’t code for proteins but regulate gene expression epigenetically
Can recruit chromatin-modifying complexes or directly interfere with mRNA stability are translation
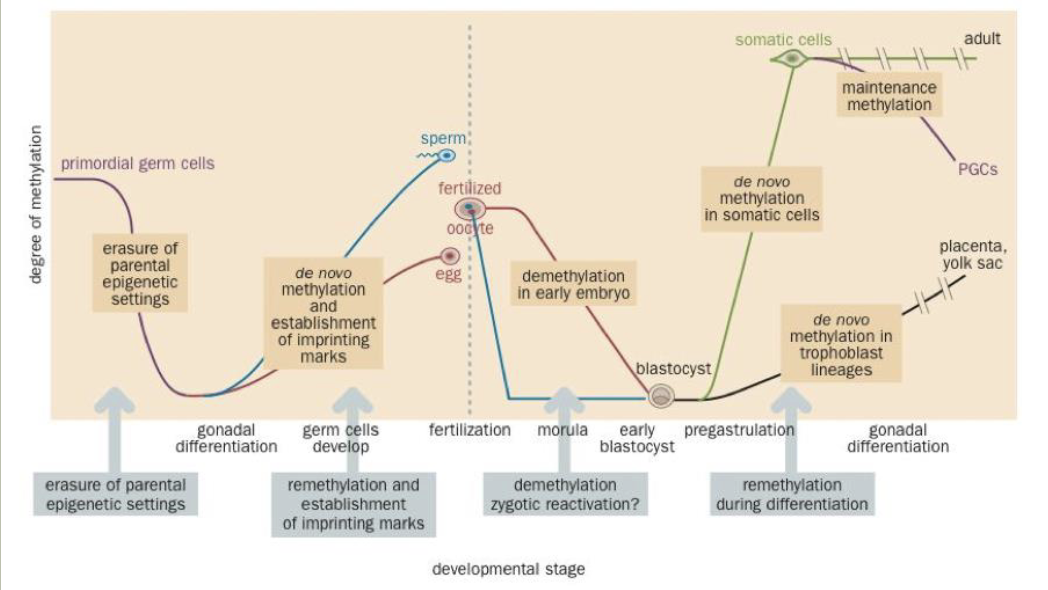
Epigenetic inheritance
The transmission of epigenetic marks (like DNA methylation) across generations without altering DNA sequence
May help adapt offspring to environmental conditions experienced by parents
Analogy: the genome as a library

Chromatin
Chromatin structure is regulated in part by histones
Euchromatin: loosely-packaged chromatin that is actively available for transcription
Heterochromatin: tightly-packaged chromatin that is not expressed; has a tendency to spread
Constituative
Chromatin structure is stable during the cell cycle
Largely composed of repeats, few genes
Facultative
Reversible heterochromatin, can be used to regular gene expression
Ex: the X chromosome in females is regulated via the formation of facultative heterochromatin (X-inactivation forming Barr bodies)
Methods for chromatin modification
Histone-modifying enzymes
Enzymes attach or remove chemical groups from histones
Non-histone proteins bind to the chemical groups to regulate gene expression
Ex: methylation
ATP—dependent remodeling complexes
Alter the location of nucleosomes
Causes formation or disassembly of nucleosomes
Actively transcribed DNA regions tend to have fewer nucleosomes
These mechanisms can interact
Histone octamer/nucleosome
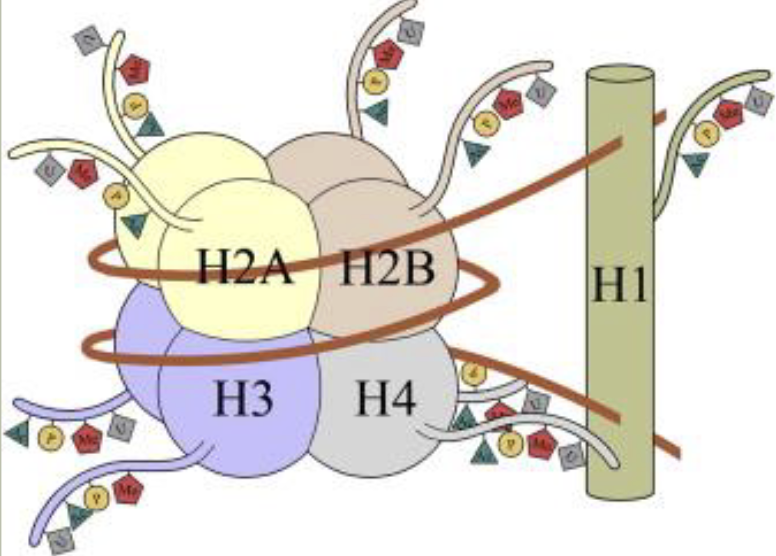
Nomenclature
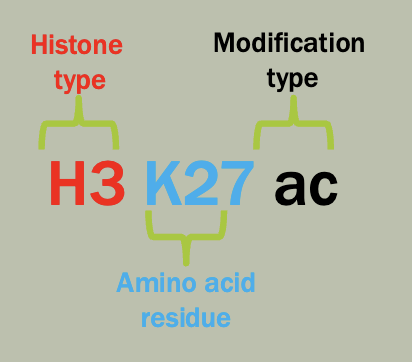
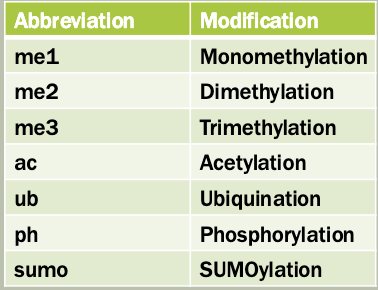
Proteins that manage and interpret post-translational modifications (PTMS) on histones
Writers: add modifications
These enzymes catalyze the addition of chemical groups (e.g. methyl, acetyl, phosphate) to histone tails
Erasers: Remove modifications
These enzymes remove the modifications effectively, reversing the writer’s work
Readers: recognize and bind modifications
These proteins or domains recognize specific histone marks and help recruit other proteins or complexes to regulate chromatin and gene expression
DNA methylation
Adding methyl groups by DNA methyltransferases to cytosine molecules that are immediately upstream of a guanine molecule (CpG sequences)
Methylation of repeat sequences is common; represses transcription; inappropriate methylation has been linked to some cancers and other diseases
Epigenetics
Lasting changes in gene expression that are not caused by changes to the DNA sequence
DNA methylation is a simple example
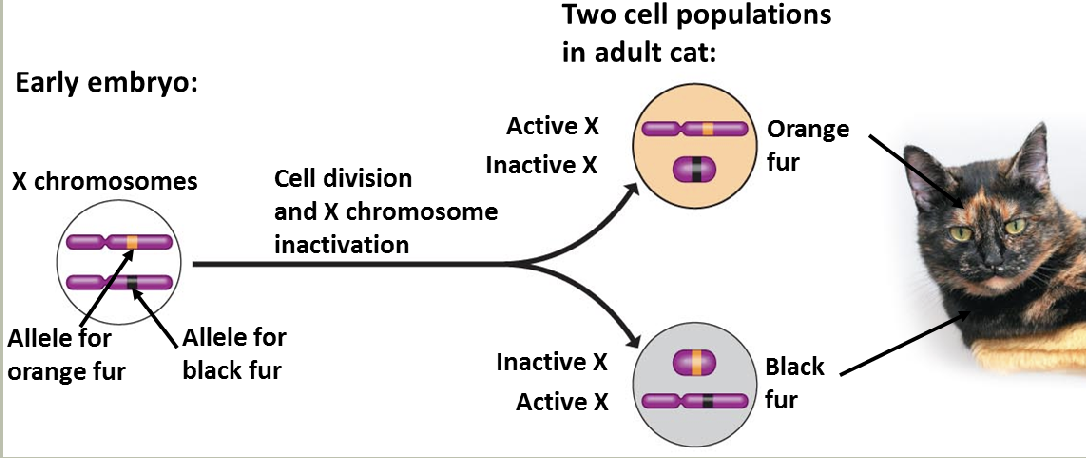
X-inactivation
X-inactivation patterns are stably transmitted from a daughter cell to its daughter cells
Not transmitted from parent to child
The X-inactivation center (XIC)
Xq13
Codes for XIST (X-inactivation specific transcript)
Produces a non-coding RNA
Recruits other proteins to inactivate the X-chromosomeIn the case of an X-autosome translocation, only the X chromosome material containing XIC deactivates, neighboring autosome material does not deactivate
Not all genes are inactive on the inactive X-chromosome
Repressed on the active X
Skewed X-inactivation
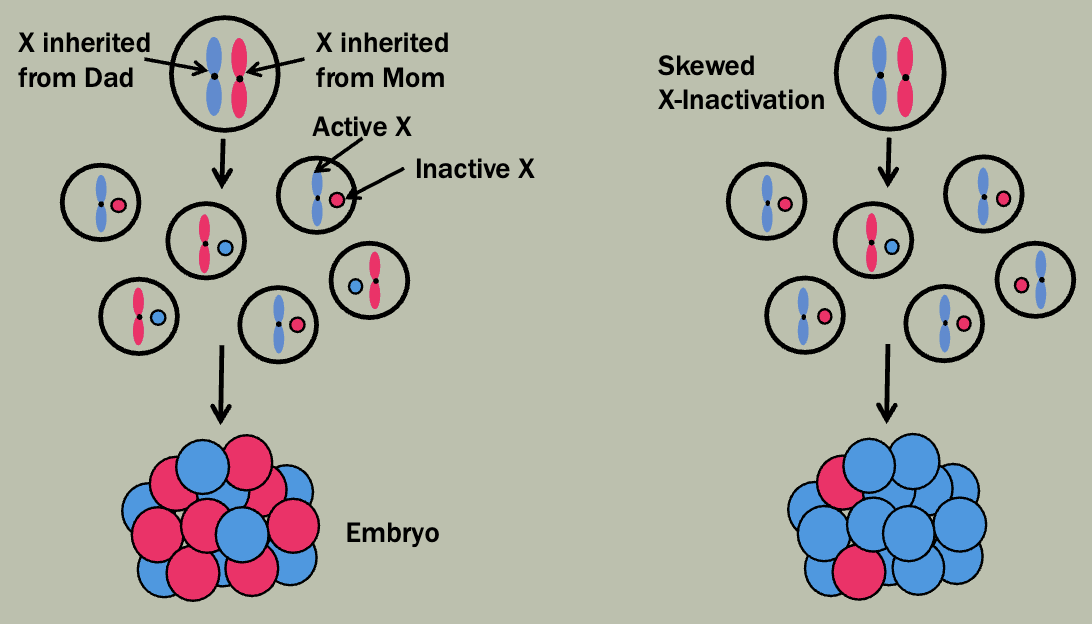
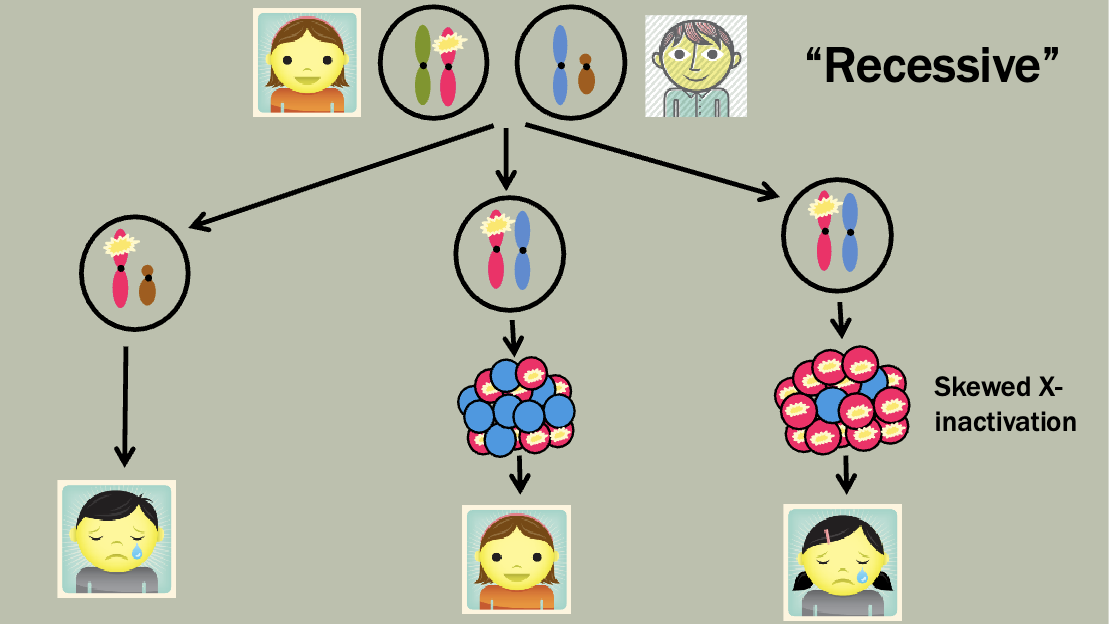
Duchenne muscular dystrophy
Dystrophin gene at Xp21 (1/4 of probands have de novo mutation)
Progressive loss of muscle function (ultimately including breathing and heart muscles
Lose ability to walk around age 9
Usually die in late teans or early 20s
Female “carriers”
Skewed X-inactivation is associated with the female phenotype
small proportion have muscle weakness and heart disease
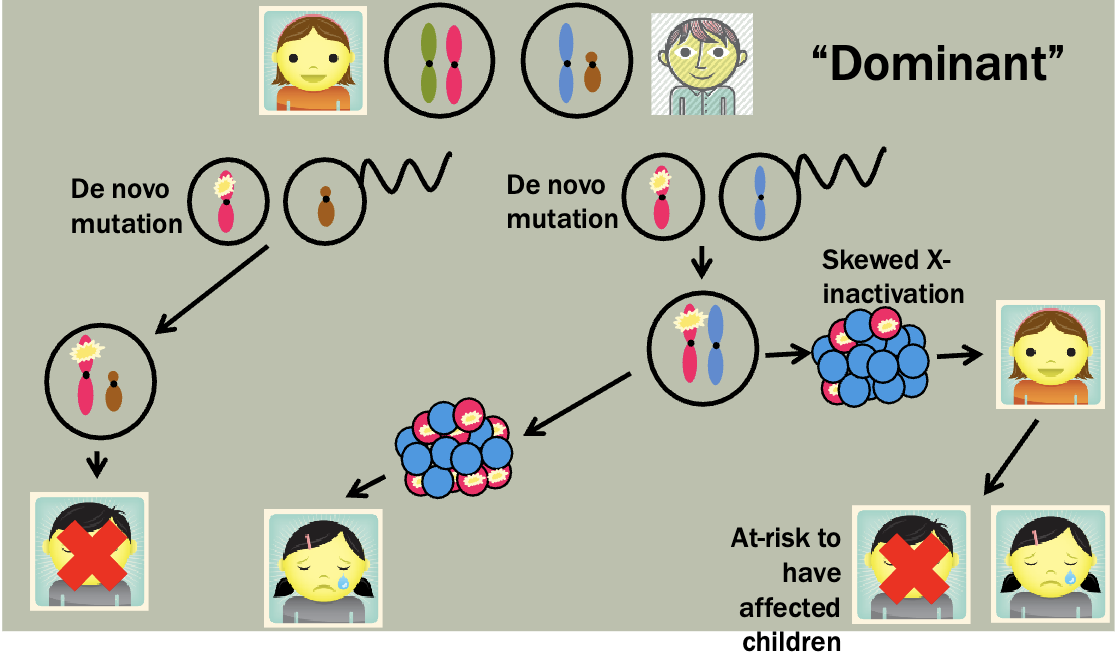
Rett syndrome
MECP2 gene at Xq28 (usually de novo)
Onset at 6-18 months in females. Lethal in males
Microcephaly and seizures
Rapid loss of developmental skills which stabilizes at 2-3 years old
highly skewed X-inactivation may result in asymptomatic women
Atypical Rett syndrome and affected alleles
Severe neonatal encephalopathy
Most common presentation in surviving males with MECP2 mutations (rare in females)
Features
Seizures
Microcephaly
Abnormal muscle tone
Breathing difficulty
Lethal by age 2
Mildly affected females and makes with classic Rett syndrome have been reported
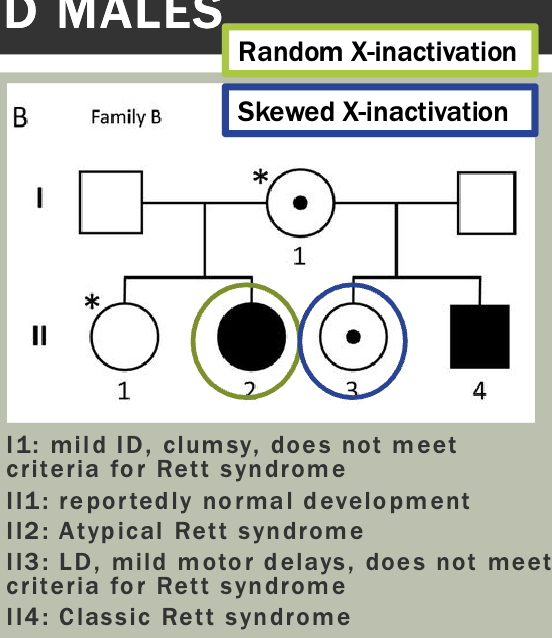
MECP2
Most mutations are de novo
Involved in neuron maturation
Expression varies across development and tissue type (differential splicing leads to brain-specific transcripts)
MECP2 protein binds to methylated CpG dinucleotides which can either activate or repress transcription
MECP2 mutations result in failure of gene silencing
Imprinting
Imprinting: gene expression is determined by the parent of origin. Imprinted genes are deactivated
Each imprinted region is likely to contain clusters of genes that are differently imprinted and some which are not imprinted at all
May be tissue-specific or developmental-stage specific
Regulated via methylation which is erased and reset shortly after conception
Chromosome 15
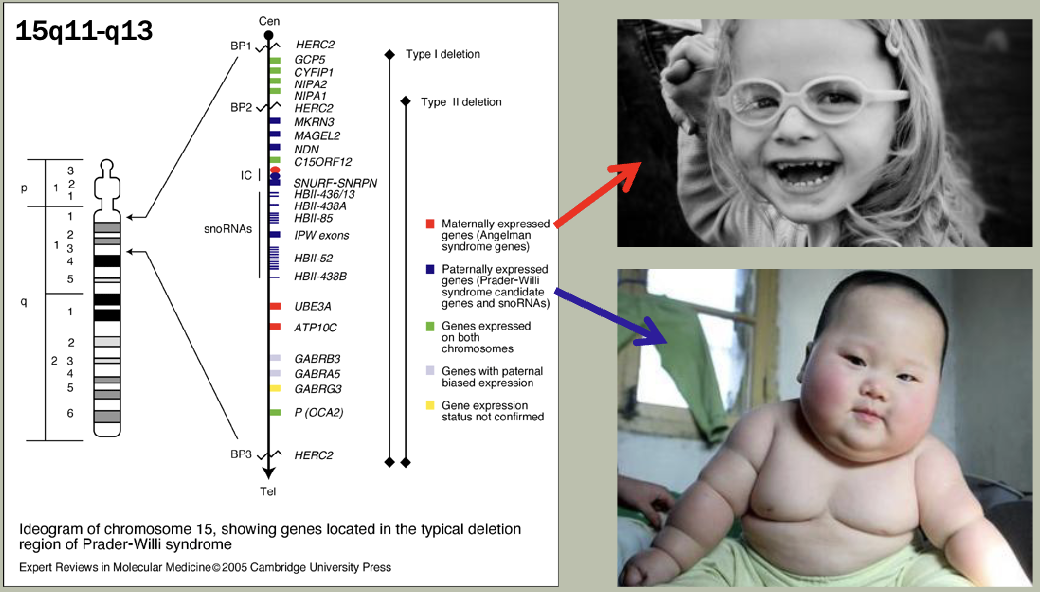
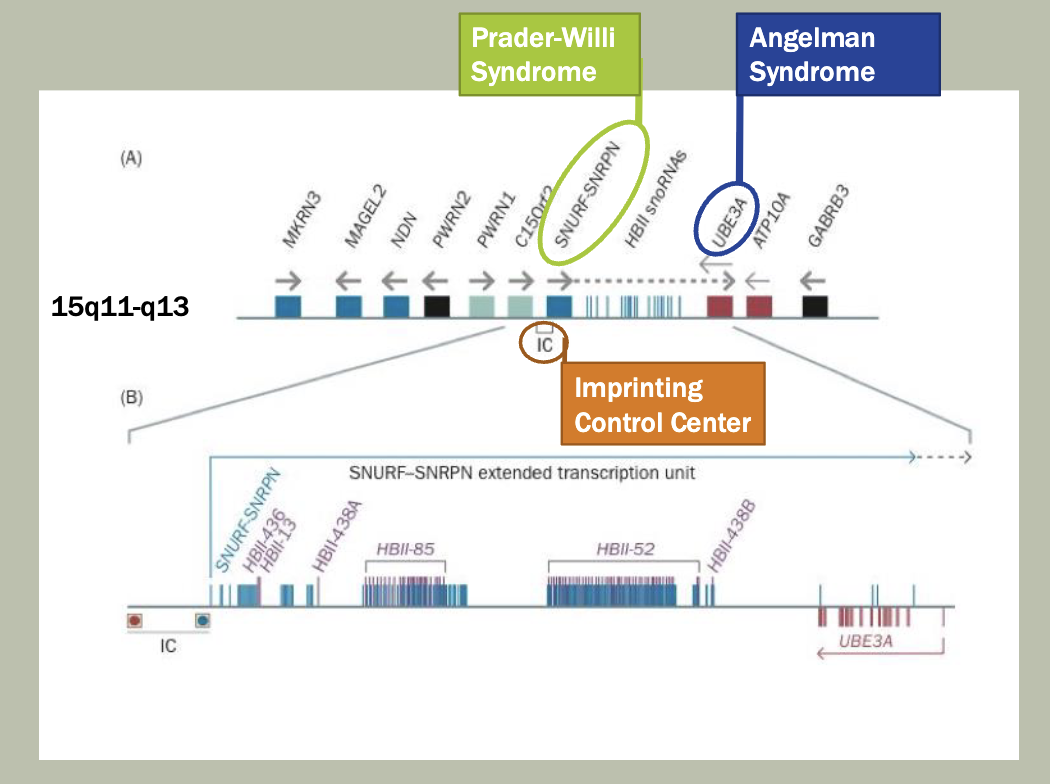
Prader-willi syndrome imprinting center (PWS-IC)
Function: controls the paternal expression of several genes in the region
Normally active on the parental allele
Regulates genes like SNRPN, SNORD116, and others that are paternally expressed
Mechanism: PWS-IC is required for establishing and maintaining the unmethylated state of the paternal allele during gametogenesis, allowing transcription of these genes
Angelman syndrome imprinting center (AS-IC)
Function: indirectly regulates the maternal expression of UBE3A by controlling methylation of the PWS-IC
Normally active on the maternal allele
Regulates imprinting and silencing of the paternal-type expression pattern in the maternal allele. Ensures UBE3A is expressed only in the maternal allele in neurons
Mechanism: THe AS-IC initiates methylation of the PWS-IC on the maternal allele during oogenesis, silencing ghte paternal-only genes there, and allowing expressing of maternal-specific genes
Summary:
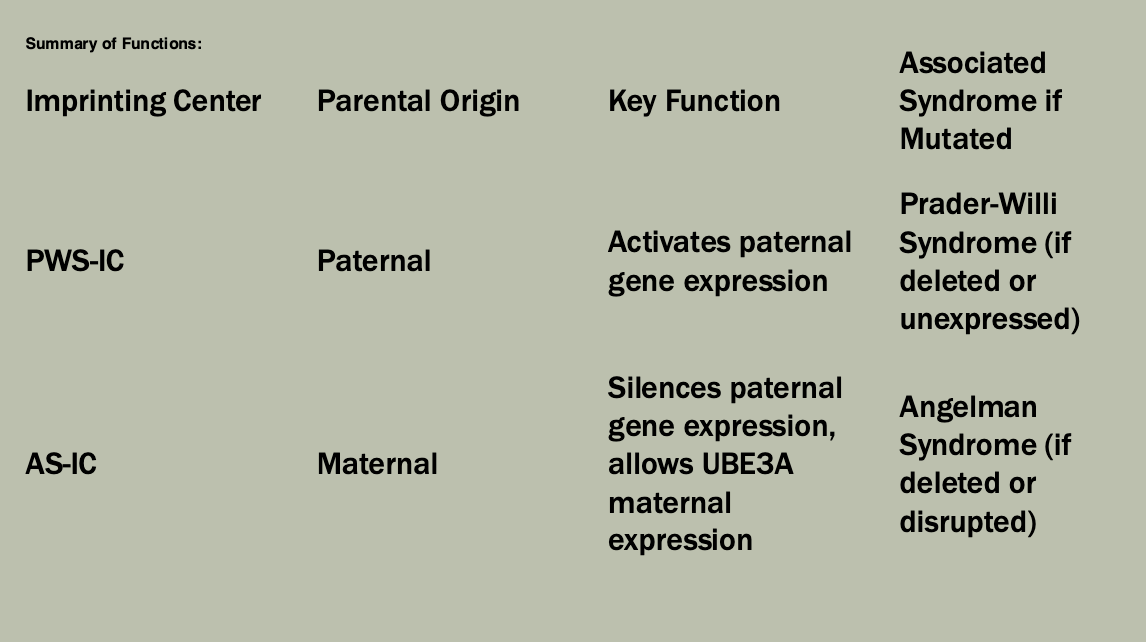
Uniparental disomy (UPD)
When two copies of the same chromosome are inherited from one parent
If the chromosome involved in the UPD is imprinted then this may cause an abnormal phenotype
Often arise via trisomy rescue
When trisomy mosaicism is noted it may be important to test for UPD
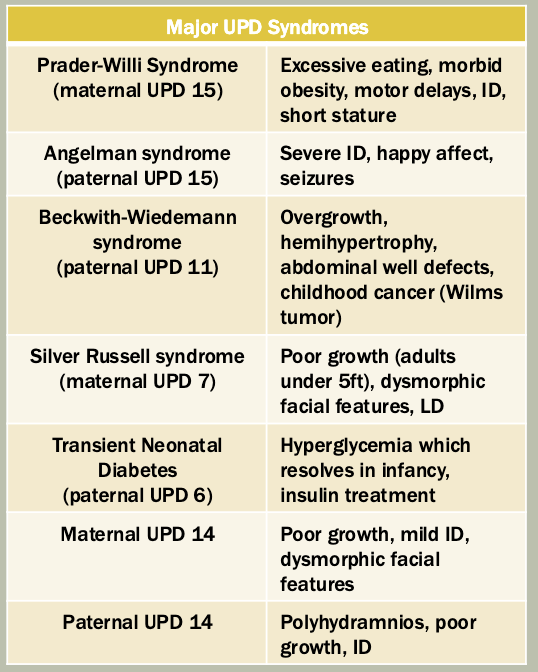
Angelman syndrome (AS)
Normal pregnancy and birth
Happy affect with inappropriate laughter, extremely social
Motor delays and severe intellectual disability
Abnormal gait
No speech
Seizures
Disrupted sleep
Normal lifespan
Maternal allele usually active!!!!

Prader-willi syndrome (PWS)
Motor developmental delay
Mild intellectual disability
Short stature
Infancy
Hypotonia
Poor appetite
Failure to thrive
Baby
Normal appetite and feeding
Normal growth
Childhood
Hyperphagia
Slow metabolism
Obesity
Parternal allele usually active!!!!
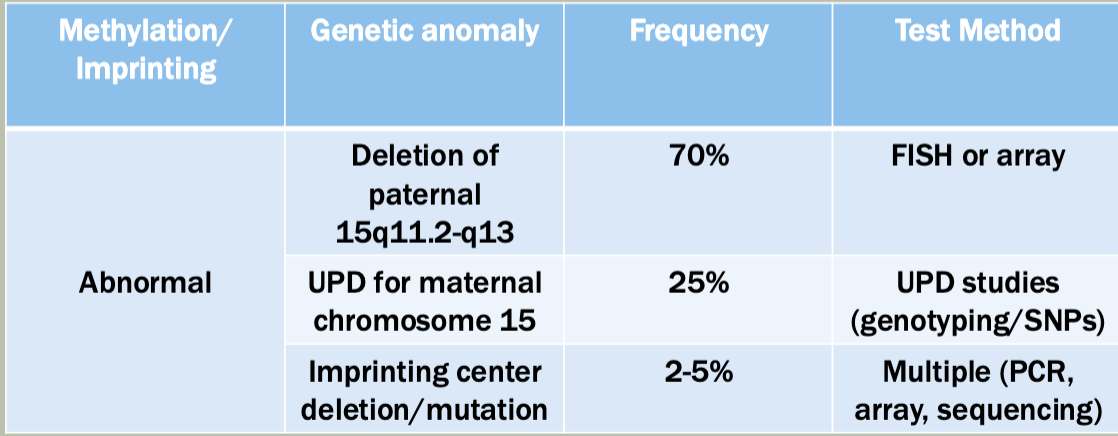
Recurring risks (for siblings)

Most cases are not inherited
Imprinting and inhertance
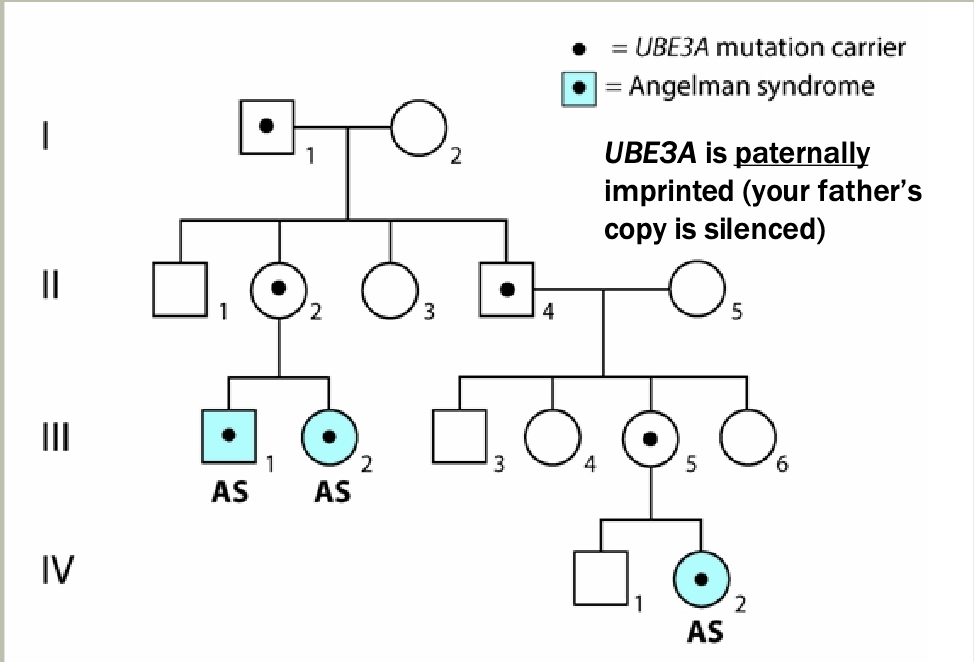
Most cases aren’t inherited, but when they are, we can study the pattern
Need to understand what is happening on the maternal and paternal chromosomes
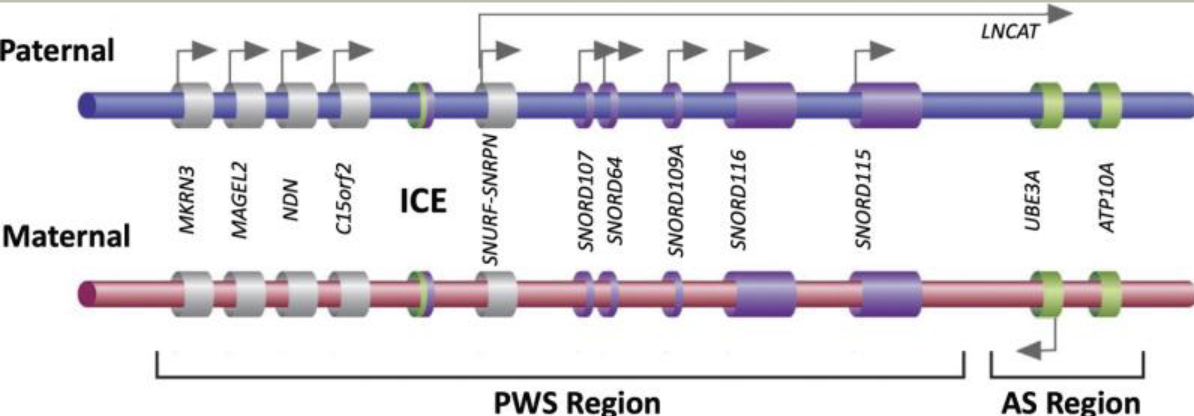
The paternal imprinting center is unmethylated, the maternal one is methylated
Paternal genes are not expressed (suppressed expression)
So this disease can be passed from mother to children, but not father to children
Key players in imprinting
What happens on paternal allele
Ice is unmethylated
Expression of SNRPN and UBE3A-ATS
UBE3A-ATS silences maternal genes in Cis
What happens on maternal allele
Ice is methylated —> silenced
UBE3A is expressed

Imprinting on a genome-wide scale
Molar pregnancy
Complete hydatidiform mole
Diploid conception with only paternal DNA present
No fetus is present
Always ends in a miscarriage
Partial hydatidiform mole
Triploid conception (69,XXX)
Usually caused by dispermy
Very abnormal placenta
Usually causes miscarriage, but rarely may result in a living child
Not all diandric triploidies cause PHM
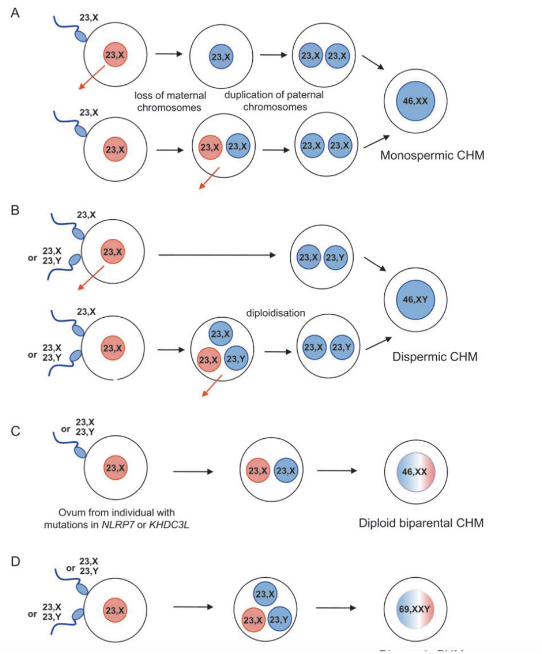
Molar pregnancies are classic examples of imprinting disorders
Complete moles have only paternal chromosomes (uniparental disomy), leading to overexpression of paternally expressed genes
Lack of maternal contribution leads to loss of maternal-imprinted gene expression, disrupting normal embryonic development
Genome-wide uniparental disomy/diploidy (GWUPD)
Case history
Baby girl born at 36 weeks (normal size)
Biliary duct cyst removed at 2 days of age
Umbilical hernia repaired at 2 months age
Kidney cyst noted but resolved spontaneously
Hearing loss diagnosed at 3 months of age
Cochlear implant at 2 years of age
GJB2 and SLC26A4 gene Sanger sequencing
negative
Homozygosity for novel V413I variant in the ESRRB
gene found (Sanger sequencing) – likely benign
Height and weight remained consistent and in the normal range through age 11 years
Mild leg length discrepancy noted
Additional genetic testing for hearing loss
Paternity confirmed using STR markers on chromosomes
Sanger sequencing of the exons of ESRRB done for family members
Whole genome SNP array fone for whole family
Results
V413I variant in the ESRRB gene found in the girl (homozygous), her brother (heterozygous) and her father (heterozygous) but NOT her mother
Paternity testing showed the girl to be homozygous at all sites for a paternal marker; no maternal markers were found
SNP array confirms genome-wide homozygosity for paternal markers
Follow-up testing og multiple tissue types showed biallelic inheritance in fibroblasts but primarily paternal inheritance in blood and saliva
Likely shows somatic mosaicism
What is the likely cause of her hearing loss?
Homozygosity for mutation in a gene causing an autosomal recessive form of hearing loss
What other conditions might she be at increased risk for?
Other individuals with similar GWUPD generally develop cancer
Hongerwinter
Lots of people were exposed to very low caloric intake
The effect from their environment affected their offspring
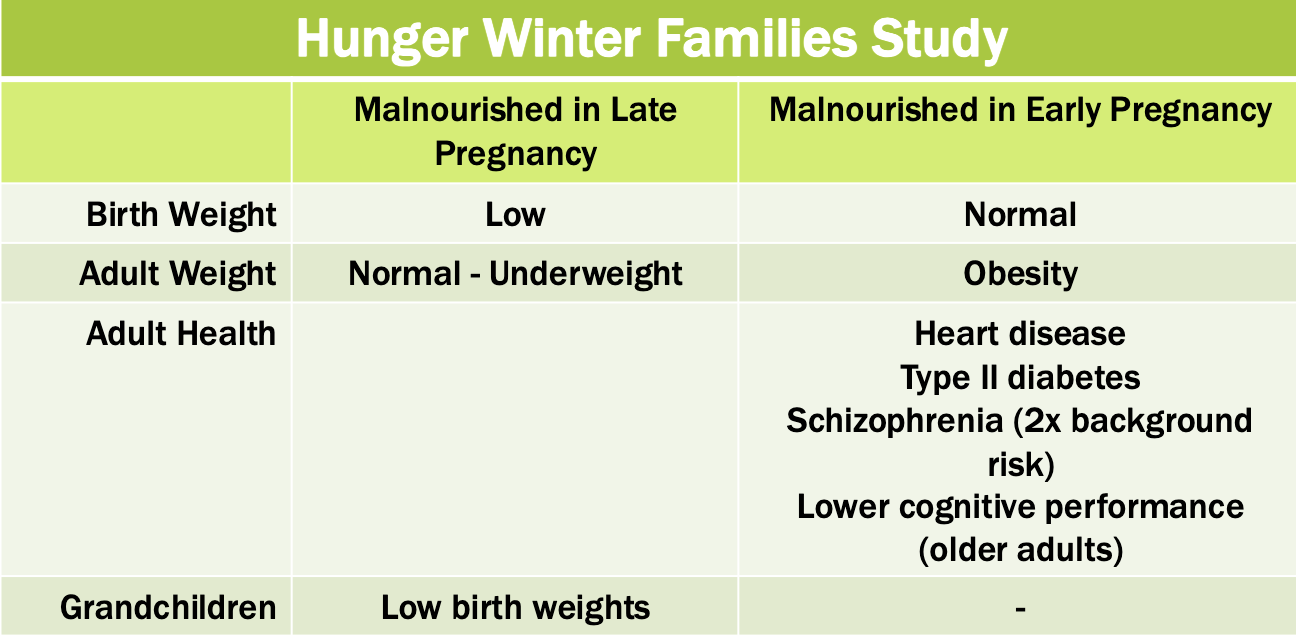
IFG2 gene — insulin-like growth factor II
11p15.5
Prenatally expressed
Regulates cellular development
Maternally imprinted (paternally expressed) except in the liver (bi-allelic)
May be associated with Wims tumor and/or Beckwith-Wiedemann syndrome
DMR: differentially methylated region
Study population: hunger winter family study
600 individuals conceived during the hunger winter
Measureed methylation of IGF2 DMR
Compared i=people with early prenatal exposure to those with late prenatal exposure
Famine during early gestation associated with hypomethylation of the IDF2 DMR
Early embryonic/fetal development may be a sensitive period for developing appropriate epigenetic markers

NASA twin study
Identical twins Mark and Scott
Scott spent one year in space
Mark stayed on earth
Study conclusions
Physiological effects of spaceflight
Stress caused by oxygen deprivation
Increased inflammation
Nutrient shift
Genetic effect of spaceflight
Telomeres lengthened while in space but returned to normal within two days of Scott’s return to Earth
Epigenetic regulation was altered during spaceflight which may highlight candidate genes related to cellular stress responses (all but 7% of these returned to normal shortly after his return
Central dogma revisited
One gene can have many promoters altering how and when
the gene is expressed
Alternative splicing can allow one gene to produce several
different protein
Post-transcriptional RNA editing alters the RNA transcript so
that the mRNA has a different sequence than the DNA
template
Environmental factors may modify epigenetic markers to
cause lasting changes to DNA
4/8/25 — recorded lecture, need to watch
put notes in flashcards!
4/10/25
took most notes in flashcards!
Cystic fibrosis
caused by mutations in the CFTR gene disrupt flow of chloride ions,
Cholera
vibrio cholerae hyperactivates the CFTR ion channels
Increased ion flow leads to more watery secretions producing the intense watery diarrhea associated with cholera
Microbiome
Environmental alterations can produce changes in microbiome composition
Composition changes over course of lifetime
4/15/2025 — also in flashcards
Case of the cranky baby
Length of pregnancy: 41 weeks
Delivery by C-section
Feeding: normal progression of feeding strategies
One sibling
Incidence of infections
Ear infections
Tubes that drain the inner ear build up, causes pressure
Can be treated with antibiotics
3+ infections warrants referral to an ENT
C.difficile
Most expensive healthcare associated infection
Contagious
Related to antibiotic use
Normally suppressed by native bacteria
High recurrence rate following treatment
Possible mechanism
Intestinal bacteria normally regulate bile
acids
Antibiotic-related loss of native bacteria may
lead to an increase in the products of bile
acid metabolism (deoxycholate)
Deoxycholate promotes the growth of C. difficile

Case study — microbes and obesity

things to consider in a fecal donor
BMI: you can’t donate for fecal transplantation if your BMI is too high
Microbiome is so tied to weight
Review
Know
types of mutations
mutations in places that disrupt splicing — cause insertions in reading frame
base pair changes
Types of variation
Methylation
TNR expansion
SNPs
Deletions/duplications
mtDNA
Nomenclature
p.F508 — most common deletion in CF
Pathogenic vs benign and everything in between
CF: everything in the slide picture
Caused by thousands of independent mutations
Shortened proteins
Sickle cell
Caused by missence mutation
Kabuki syndrome
KMT2D (12q13.12) and KDM6A (Xp11.2)
GWAS
4/24/2025
Evolution basis
Multi-regional model (Thorn and Wolpoff
Modern HUmans evolved in multiple regions from Homo erectus
Based on fossil evidence linking ancient and modern populations
Out of Africa model
Modern humans evolved in Africa and dispersed recently
Based on genetic evidence
Major events in the study of human evolution
Discovery of DNA structure
Development of molecular clock hypothesis
Proposed that mutations accumulate at a roughly constant rate. Enabled estimation of divergence times between species
Mitochondrial Eve and Out-of-Africa hypothesis
Analyzed mitochondrial DNA from global populations and concluded that all modern humans share a common maternal ancestor who lifed in Africa
Y-chromosome Adam
Studies Y-chromosome variation revealed a common paternal ancestor, supporting African origin of humans
Completion of the human genome project
enabled comparative studies with other hominins and primates, revealing what makes humans unique
Sequencing of Neanderthal genome
Demonstrated interbreeding between Neanderthals and modern humans
Advances in Ancient DNA techniques
Populartion genomics and modern human diversity
CHISPR and functional genomics
Mitochonrial eve
Most recent common female ancestor from which all modern humans descended
Comparative analysis of mtDNA variation suggests that a single woman who lived ~200,000 years ago is the ancestor of all humans alive today
Showed that all lineages recently trace back to Africa
Y-chromosome Adam
Most recent common male ancestor from which all modern humans descended
Comparative analysis of Y chromosome variation suggests that a single man who lived 180-200,000 years ago
1000 genomes project
Catalogue of human genetic variation across many population'
Sequenced the genomes of 2504 people across 26 populations
Low coverage WGS
Deep exome sequencing
Dense microarray analysis
Lacks full representation of some global regions
How does 23 and me work?
Genotyping your DNA
Comparing to reference panels
Statistical algorithms
Output: your ethnicity estimate
Important caveats
Estimates evolve
Geographic labels are fuzzy
Not suitable for detailed ethnic identification
More accurate for recent ancestry
Neanderthals
First discovered in 1856
Neanderthals left Africa 500,000 years ago and migrated to Europe and Asia before modern humans
Largely limited to Europe
Went extinct 30,000 ya
Complex societies
Neanderthal DNA
Alleles contribute to behaviors
Chronotype
Tendency towards loneliness and isolation
Frequency of lack of enthusiasm and disconnect
Smoking statuesnyt
Neanderthal dessert: area of a genome without neanderthal DNA
We think that humans had complex language, but neanderthals didn’t
FOXP2
7q31.1
Regulates the brain’s control of the lips, mouth, and tongue
Critical for normal language development
FOXP2-related speech nd language disorder
Apraxia of speech
Sickle cell anemia
5 haplotypes associated with hemoglobin S muation
ATA
2003 skeleton found in Chile
6 inches big
Mitochondrial B2 haplotype matched to South American populations
bone age suggested 6-8 years of age.
2 X chromosomes - predicted to be female
Multiple SNVs found associated with skeletal phenotypes
April 29 2025
Ethical issues in human genetics
Pace of tech change is faster than society’s ability to comprehend and adapt
Genotyping and disease prediction is misunderstood
Porential benefits are being missed
New tools of genetic modification, forensic testing, etc. are already here
Testing
A test can have implication for other family members
A person has, at some level, the right to not know
Is there a duty of care?
If you are a practitioner and you know that someone has a disorder, do you have to tell them?
Huntington disease
No cure or effective treatment
Genetic testing is available and extremely accurate
75% of individuals at HD risk choose not to get tested
Key features that lead to ethical dilemma
autosomal dominant (50% risk to offspring)
anticipation (getting worse or appearing earlier in future generations) in some cases
No cure
Testing deeply affects identity
Disclosure to at-risk relatives
Most genetic tests have serious limitations
Most have limited predictive power
Might know that the condition will exist but not how severe it will be
Might find a disease that they weren’t expecting to find
Potential for discrimination
Particularly with presymptomatic testing
Laws protecting us are limited and subject to change
Testing and reproductive choices
Children are a major issue for many people
The effect of parents’ genetic status complicated things
Termination vs preparation are major decisions with very strong social and cultural ramifications
Direct to consumer testing
Limited state and federal regulation
Questionable clinical value
not all insurance concerns are addressed
privacy policies not backed by laws
no regulation of genetic variants that can be included in risk assessments
increases global health inequality
Inappropriate genetic testing
Sex selection
Paternity
Generally requires establishing legal chain of custody to be used in court in cases of
custody
child support
inheritance claims
immigration
adoption
Forensic testing generally done a pre-existing samples
Forensic DNA databases
CODIS
uses only DNA from people with convictions
Genetic genealogy (gedmatch)
Works better than CODIS
Open database
More likely to get a match to a family member
Appropriate uses
Potential for error