24-Biochemistry-Lecture24 | Week 13 - Lecture 2
Nitrogen Metabolism and Urea Cycle
Lecture Overview: Nitrogen Metabolism
Key Topics:
Digestion of proteins in animals
Degradation of amino acids in animals
Synthesis and excretion of urea
Metabolic Circumstances of Amino Acid Oxidation
Amino acids undergo oxidative degradation under certain conditions:
When released during protein turnover, if not needed for new protein synthesis.
When ingested amino acids exceed the body's protein synthesis needs.
When cellular proteins are used as fuel due to lack of carbohydrates (e.g., starvation, diseases such as diabetes).
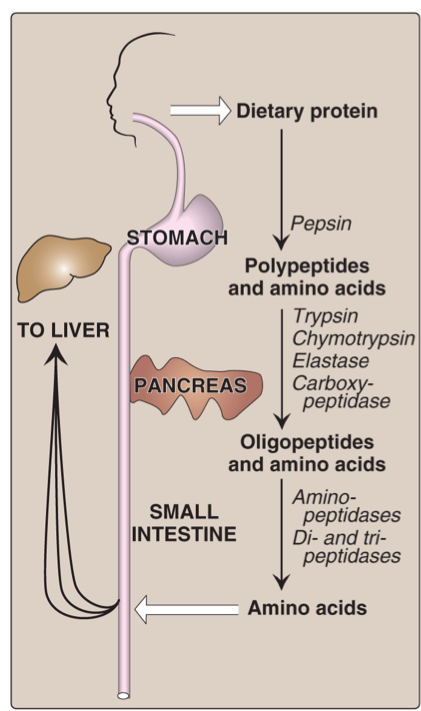
Dietary Protein Digestion
Proteins are enzymatically hydrolyzed into amino acids:
Stomach: Pepsin degrades proteins into large peptides.
Small Intestine: Further breakdown by enzymes including trypsin, chymotrypsin, elastase, aminopeptidase, and carboxypeptidases into smaller peptides and amino acids.
Amino acids and small peptides are transported into blood and then to the liver.
intestine lining cells → blood → liver
Overview of Amino Acid Catabolism
Amino Group: Enters the urea cycle and nitrogen is excreted as urea.
Carbon Skeleton: Enters the citric acid cycle for oxidation or energy use.
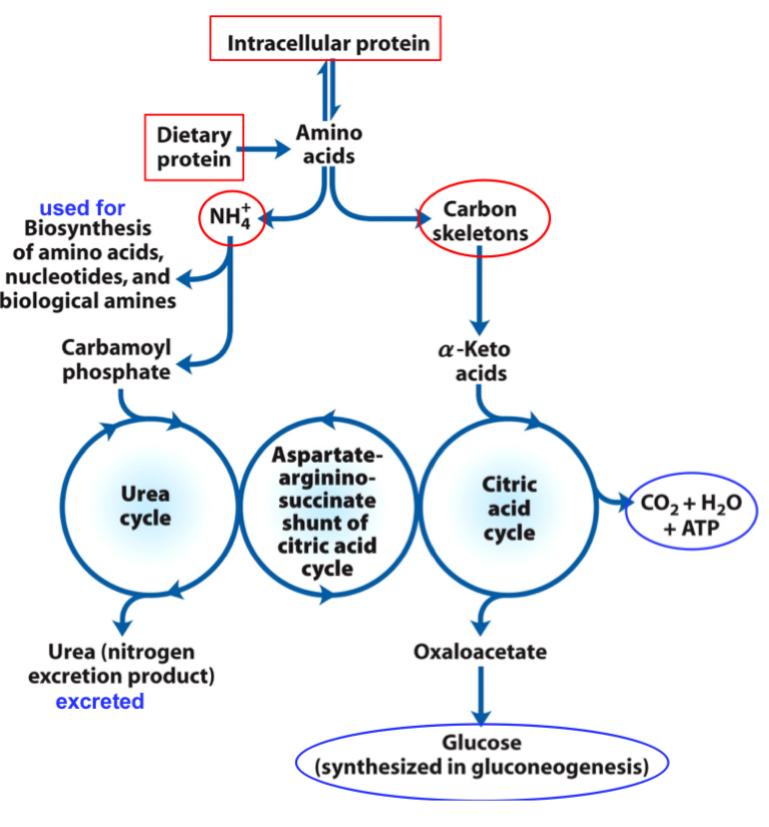
Metabolic Fates of Amino Groups
Amino Group Catabolism
The amino groups, unless reused, contribute to the formation of:
Urea (excreted)
Uric acid (derived from purines)
Ammonia (NH3) is toxic; urea is significantly less toxic and highly soluble.
Urea is far less toxic than ammonia & has very high solubility
Uric acid is water-insoluble, facilitating water conservation via paste-like excretion.
unless re-used, amino groups are channeled into a single excretory end product:
Humans and great apes excrete both urea (from amino acids) and uric acid (from purines)
THE AMINO GROUP IS TRANSFERRED TO a-KETOGLUTARATE FORMING L-GLUTAMATE
Transamination is catalyzed by aminotransferases (transaminases) and requires the cofactor pyridoxal phosphate (PLP).
An amino group from an L-amino acid is transferred to α-ketoglutarate, forming L-glutamate and an α-keto acid.
Glutamate acts as a temporary nitrogen storage molecule, carrying the amino group.
Glutamate can later donate the amino group for urea formation or amino acid biosynthesis.
The original amino acid is converted into its corresponding α-keto acid.
Transaminases
Cells contain many different Transaminases (aminotransferases).
They are specific for α-ketoglutarate as the amino group acceptor, but differ in their specificity for the amino group donor.
Named for the amino group donor, e.g.
Alanine aminotransferase transfers an amino group from alanine to α-ketoglutarate → forms pyruvate and glutamate.
Aspartate aminotransferase transfers an amino group from aspartate to α-ketoglutarate → forms glutamate and oxaloacetate.
The reactions catalyzed by transaminases are reversible.
All amino transferases use pyridoxal phosphate (PLP) as a prosthetic group.
This is the coenzyme form of pyridoxine, or vitamin B₆.
Pyridoxal phosphate is tightly bound to the transaminase active site through a Schiff-base (covalent) and other non-covalent interactions.
PLP IS COVALENTLY LINKED TO TRANSAMINASES
PLP (pyridoxal phosphate) is covalently linked to transaminases by an internal aldimine.
The linkage forms via nucleophilic attack by the amino group of an active-site lysine.
In the enzyme, PLP binds tightly to the active site of aminotransferases like aspartate aminotransferase.
STRUCTURE OF PLP AND PYRIDOXAMINE PHOSPHATE
PLP acts as an intermediate, enzyme-bound carrier of amino groups.
The aldehyde form (PLP) reacts reversibly with amino groups.
The aminated form (pyridoxamine phosphate) reacts reversibly with carbonyl groups.
PLP FUNCTIONS AS AN INTERMEDIATE CARRIER OF AMINO GROUPS AT THE ACTIVE SITE OF TRANSAMINASES
PLP undergoes reversible transformation between its internal aldimine form (pyridoxal phosphate), which accepts an amino group, and its aminated form (pyridoxamine phosphate), which donates the amino group to a keto acid.
This transformation occurs through a Schiff base intermediate.
In the reverse reaction, a second α-keto acid replaces the first and is converted to an amino acid.
PLP also participates in reactions at the β and γ carbons of some amino acids.
TRANSAMINASE LEVELS IN SERUM CAN INDICATE TISSUE DAMAGE
Transaminase (aminotransferase) levels in serum are used to assess tissue damage in the heart, liver, and pancreas.
When tissues are damaged, enzymes leak into the bloodstream, where they can be detected.
Timing and enzyme specificity help determine the severity and origin of the injury.
The most common tests measure:
sGOT – serum Glutamate-Oxaloacetate Transaminase (aka AST/aspartate aminotransferase)
sGPT – serum Glutamate-Pyruvate Transaminase (aka ALT/alanine aminotransferase)
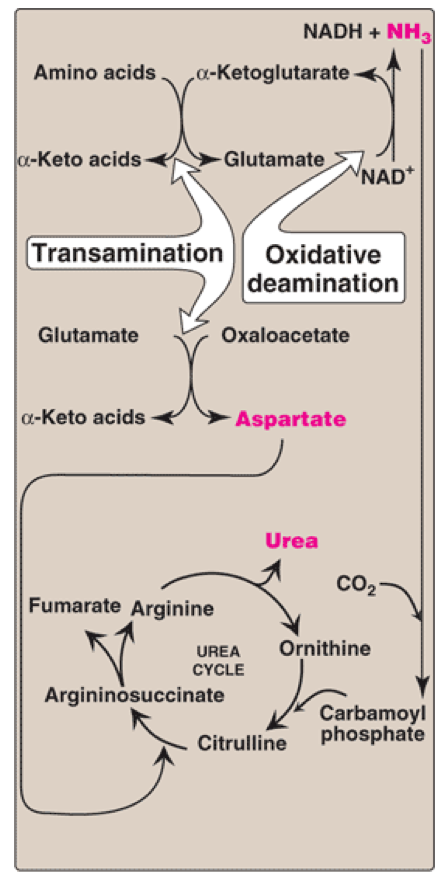
IN THE LIVER, AMMONIA COLLECTED IN GLUTAMATE IS REMOVED BY GLUTAMATE DEHYDROGENASE (GDH) TO BE EXCRETED AS UREA
In hepatocytes (liver), glutamate is transported from the cytosol to mitochondria.
Glutamate undergoes oxidative deamination by glutamate dehydrogenase (GDH) in the mitochondrial matrix, producing α-ketoglutarate and releasing ammonia (NH₄⁺).
GDH can use either NAD⁺ or NADP⁺ as the electron acceptor.
Released ammonia is converted into urea for excretion.
α-Ketoglutarate can enter the citric acid cycle or be used for gluconeogenesis.
This process is part of transdeamination, which combines transamination (by transaminases) and oxidative deamination (by GDH).
Glutamate Dehydrogenase (GDH) in Carbon & Nitrogen Metabolism
Nitrogen metabolism: Ammonia is produced and converted to urea for excretion.
Carbon metabolism: Produces α-ketoglutarate, which can be:
Oxidized in the citric acid cycle (CAC)
Used for gluconeogenesis (via oxaloacetate)
GDH is allosterically regulated:
Activated by ADP (low energy → promotes amino acid degradation for energy)
Inhibited by GTP (high energy/CAC activity → suppresses amino acid degradation)
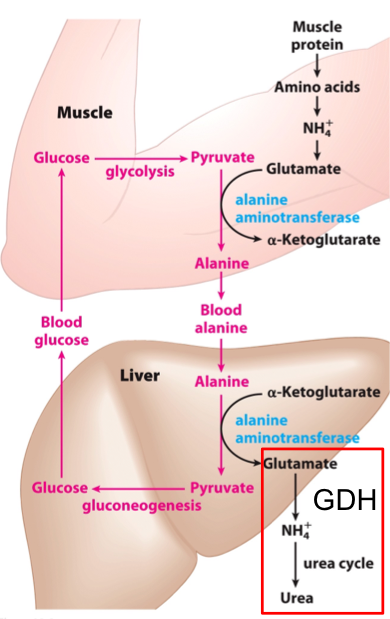
In the Muscle, Ammonia Collected in Glutamate is Donated to Pyruvate to Make Alanine
Working muscles use glycolysis → producing pyruvate and operating anaerobically.
If pyruvate accumulates, it can lead to lactic acid buildup.
To prevent this, pyruvate accepts an amino group from glutamate, forming alanine for transport to the liver.
In the liver, alanine donates its amino group → becomes pyruvate → converted to glucose.
Glucose returns to the muscle; ammonia is excreted (via the glucose-alanine cycle).
Alanine Transports Ammonia From Skeletal Muscle to the Liver: The Glucose-Alanine Cycle
Alanine plays a key role in transporting amino groups to the liver via the glucose-alanine cycle.
In this cycle, the amino group from glutamate is transferred to pyruvate by alanine aminotransferase, forming alanine in muscle.
Alanine is transported through the blood to the liver.
In the liver, alanine is transaminated back to pyruvate and glutamate.
Pyruvate undergoes gluconeogenesis to form glucose, which is sent back to the muscle.
Ammonia from the deamination of glutamate (via GDH) enters the urea cycle and is excreted.
AMMONIA FROM OTHER TISSUES IS SAFELY TRANSPORTED TO THE LIVER IN THE BLOODSTREAM AS GLUTAMINE
Many tissues, including the brain, produce free ammonia, which is toxic and must be transported in a non-toxic form.
Excess ammonia in tissues is added to glutamate to form glutamine, via glutamine synthetase (requires ATP).
Glutamine is transported in the bloodstream to the liver.
In the liver mitochondria, glutaminase releases ammonia (NH₄⁺) from glutamine, regenerating glutamate.
The liberated NH₄⁺ enters the urea cycle for excretion.
Excess glutamine is also processed in the intestines, kidneys, and liver.
Toxic Effects of Ammonia
NH₃ crosses the blood-brain barrier and accumulates in brain cells.
Free ammonia is especially toxic to the brain: causes cognitive impairment, ataxia, seizures, swelling, and potentially death.
NH₄⁺ competes with K⁺ for transport into astrocytes via Na⁺/K⁺ ATPase.
Increases extracellular K⁺ in neurons → triggers excess K⁺ influx via NKCC1 (Na⁺-K⁺-2Cl⁻ symporter).
Excess Cl⁻ alters GABAergic signaling, leading to neuromuscular incoordination and seizures.
The Fate of Ammonia: The Urea cycle & Nitrogen Excretion
HIGHLY TOXIC AMMONIA MUST BE UTILIZED OR EXCRETED IN UREA
Urea excretion mechanism → urea contains 2 amino groups
First nitrogen from free ammonia; catalyzed by carbamoyl phosphate synthase I → captures the free ammonia in the mitochondrial maatrix
Second nitrogen from aspartate via argininosuccinate synthetase.
The second amino group of urea is acquired from aspartate
Requires ATP for activation.
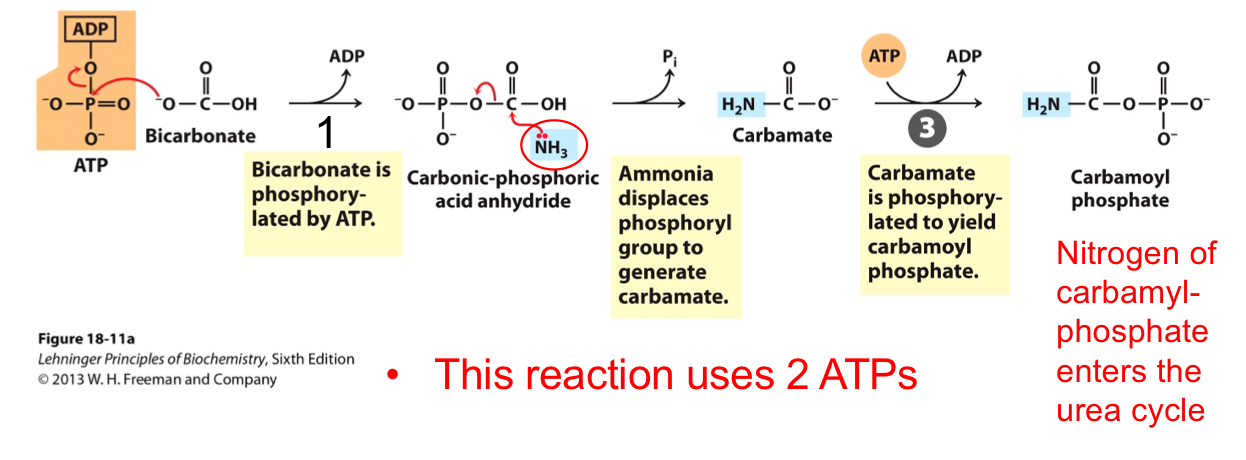
THE FIRST NITROGEN-ACQUIRING REACTION OF THE UREA CYCLE
(Synthesis of carbamoyl phosphate from ammonia)
Catalyzed by carbamoyl phosphate synthetase I
Nitrogen enters from free ammonia (NH₄⁺)
2 ATP are used
This reaction has 2 activation steps
Carbamoyl phosphate enters the urea cycle
THE SECOND NITROGEN-ACQUIRING REACTION OF THE UREA CYCLE
(Entry of aspartate into the urea cycle)
Catalyzed by argininosuccinate synthetase
Aspartate donates the second nitrogen
Forms argininosuccinate
2 ATP equivalents used (1 ATP → AMP + PPi)
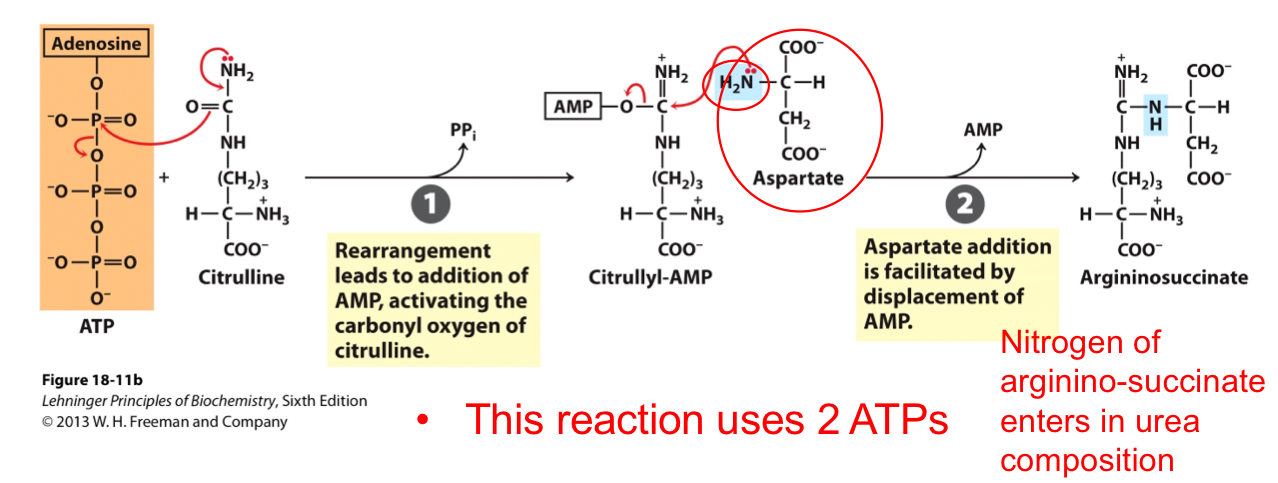
THE AMINO GROUP OF GLUTAMATE OR GLUTAMINE IS METABOLIZED IN THE MITOCHONDRIA OF HEPATOCYTES
Glutamate or glutamine releases free NH₄⁺
Carbamoyl phosphate synthetase I captures free NH₄⁺ in mitochondria
First step of the urea cycle
Regulated
Two nitrogen sources for urea:
NH₄⁺ (from glutamate or glutamine)
Aspartate (from oxaloacetate via transamination)
THE REACTIONS IN THE UREA CYCLE
Enzymes are located in both mitochondria and cytosol
Two amino groups enter:
Carbamoyl phosphate
Aspartate
4 major steps in the urea cycle (3 are irreversible):
Citrulline formation from ornithine and carbamoyl phosphate (Ornithine transcarbamylase)
Argininosuccinate formation from citrulline and aspartate (Argininosuccinate synthetase)
Arginine formation from argininosuccinate (Argininosuccinate lyase)
Urea formation and ornithine regeneration (Arginase)
ATP COST OF THE UREA CYCLE
4 ATP equivalents are required:
2 ATP for carbamoyl phosphate synthesis
1 ATP → AMP + PPi in argininosuccinate formation (cost = 2 ATP equivalents)
Total = 4 ATP (or equivalents)
Some energy is recovered:
Fumarate is converted to malate, feeding the CAC
Additional recovery from GDH reaction
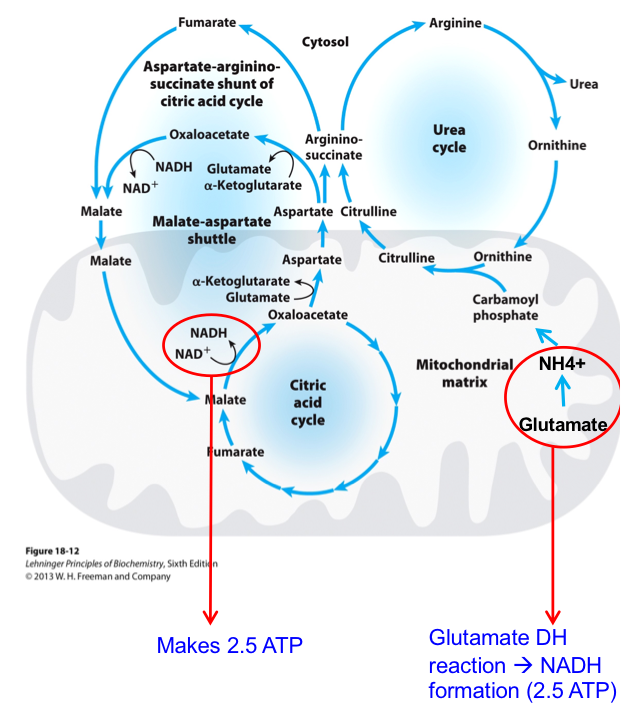
The Urea Cycle and Citric Acid Cycle Are Linked (Krebs Bicycle)
The aspartate-arginosuccinate shunt links the urea and citric acid cycles.
It connects amino acid nitrogen disposal to energy metabolism.
NADH is produced (2.5 ATP) in malate to oxaloacetate conversion and by reoxidation of fumarate.
Glutamate dehydrogenase reaction produces NADH for ATP → 2.5 ATP molecules
Total: 5 ATP made
Thus, the urea cycle can be considered self-sustaining.
Review: Flow of Nitrogen from Amino Acids to Urea
Overall reaction:
Aspartate + NH₄⁺ + HCO₃⁻ + 3 ATP + H₂O → Urea + fumarate + 2 ADP + AMP + 2 Pi + PPi
PPi → 2 Pi = 1 ATP equivalent
Thus, 4 ATP are used
Urea is mainly excreted in the urine.
Hyperammonemia occurs if the liver function is compromised or urea cycle enzymes are deficient.
NH₄⁺ > 100 μM is toxic.
Mechanisms may include:
Depletion of α-ketoglutarate → ↓ TCA cycle activity
↓ neurotransmitters like glutamate and GABA
Regulation of the Urea Cycle by N-Acetylglutamate and Arginine
N-Acetylglutamate is formed by N-acetylglutamate synthase.
Formed when glutamate and acetyl-CoA are high.
N-acetylglutamate activates carbamoyl phosphate synthetase I (CPS I).
High arginine levels increase N-acetylglutamate synthesis.
Gene expression of urea cycle enzymes increases with:
High-protein diet
Starvation (when protein is broken for energy)
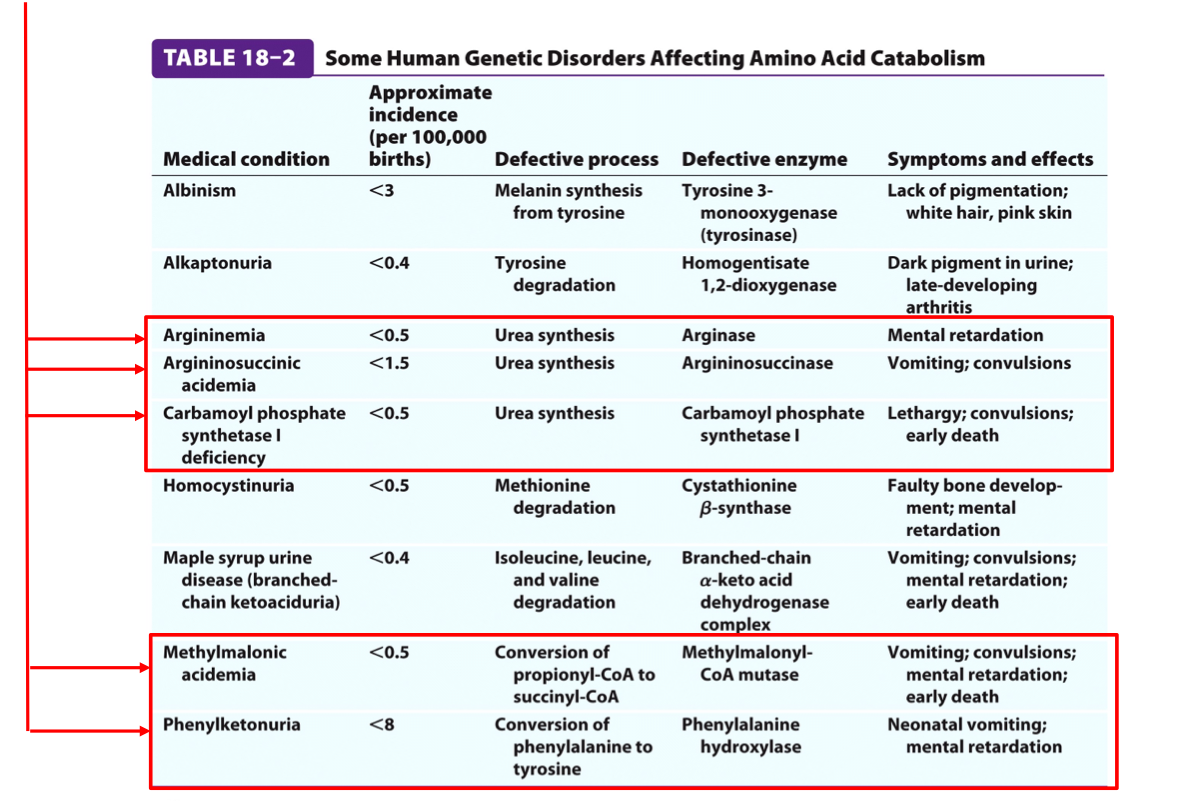
Genetic Defects in the Urea Cycle Can Be Life-Threatening (JC10)
Individuals with urea cycle enzyme deficiencies cannot tolerate high-protein diets.
Ammonia is highly toxic:
Amino acids are deaminated → ammonia builds up → neurotoxic.
Deficiency in any enzyme → hyperammonemia and toxic buildup of intermediates.
Genetic screening identifies which intermediate is accumulating.
Humans cannot synthesize ~50% of amino acids → must obtain from diet.
JC10: Genetic Defects in the Urea Cycle
Possible Treatments of Defects in the Urea Cycle Enzymes
Phenylbutyrate in the diet:
Lowers ammonia levels in the blood.
Promotes urinary excretion of glutamine as phenylacetylglutamine.
Benzoate in the diet:
Lowers ammonia levels in the blood.
Promotes urinary excretion of glycine as benzoylglycine (hippurate).
Subsequent synthesis of glycine and glutamine helps replenish their pools and further removes ammonia from the bloodstream.
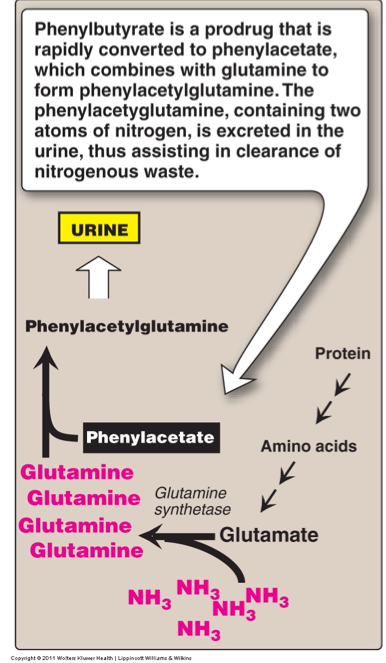
Mechanism of Action of Phenylbutyrate and Benzoate
Benzoate:
Converted to benzoyl-CoA using ATP.
Benzoyl-CoA reacts with glycine to form benzoylglycine (hippurate), which is excreted in the urine.
The glycine used must be regenerated, which removes ammonia.
Phenylbutyrate:
Undergoes β-oxidation → phenylacetate.
Phenylacetate + CoA + ATP → phenylacetyl-CoA.
Phenylacetyl-CoA reacts with glutamine → phenylacetylglutamine, which is excreted in urine.
These processes lower blood ammonia by removing nitrogen via glycine and glutamine.
Severe Hyperammonemia Treatment
Treated by hemodialysis in emergency cases.
Enzyme-Specific Therapies
N-acetylglutamate synthetase deficiency:
No production of N-acetylglutamate (activator of CPS I).
Treated by carbamoyl-glutamate, an analog that activates carbamoyl phosphate synthetase I.
Arginine supplementation:
Used for enzyme deficiencies in steps 1, 2, or 3 of the urea cycle.
Arginine helps capture ammonia from carbamoyl phosphate or aspartate.
Summary
The urea cycle links the metabolism of amino acids and their nitrogenous wastes, while regulatory mechanisms and potential interventions can mitigate the effects of genetic defects.