RFLPs, AFLPs and Sanger sequencing
RFLP
Restriction fragment length polymorphism
Use restriction enzymes to cut DNA at short specific sequences
restriction enzymes: from bacteria, cleave foreign DNA (the bacterial DNA doesn’t get cut)
adding 1 or 2 restriction enzymes to purified dna turns a single piece of dna into multiple fragments
some individuals may only have one restriction site whereas others have two or three: the resulting fragments are different lengths
AFLP
amplified fragment length polymorphism
pcr-based
often used for plants
amplificaion of multiple fragments = diffrent-sized bands on a gel
pattern of bands depends on sequences immediately adjacent to adaptors and distance between restriction sites
Sanger sequencing
Transcription Translation
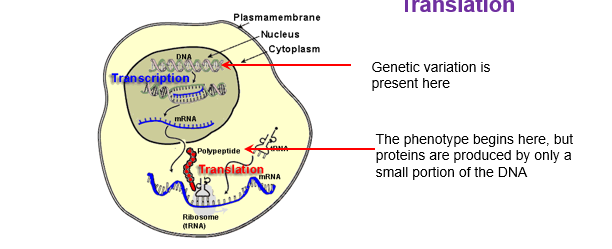
The sequence of base pairs in dna will determine sequence in the mRNA
three base pair sequences on mRNA are called codons
Uracil in RNA replaces thymine in DNA
Translation:
the sequence of bases in the codon determines the sequence of amino acids in the protein
redundancy in the genetic code means that many substitutions in DNA do not result in altered proteins (synonymous substitution)
ex. CUU, CUC, CUA, CUG all code for leucine
Junk DNA vs genes that code for products: Exons are transcribed, introns are not Most variations is in the introns
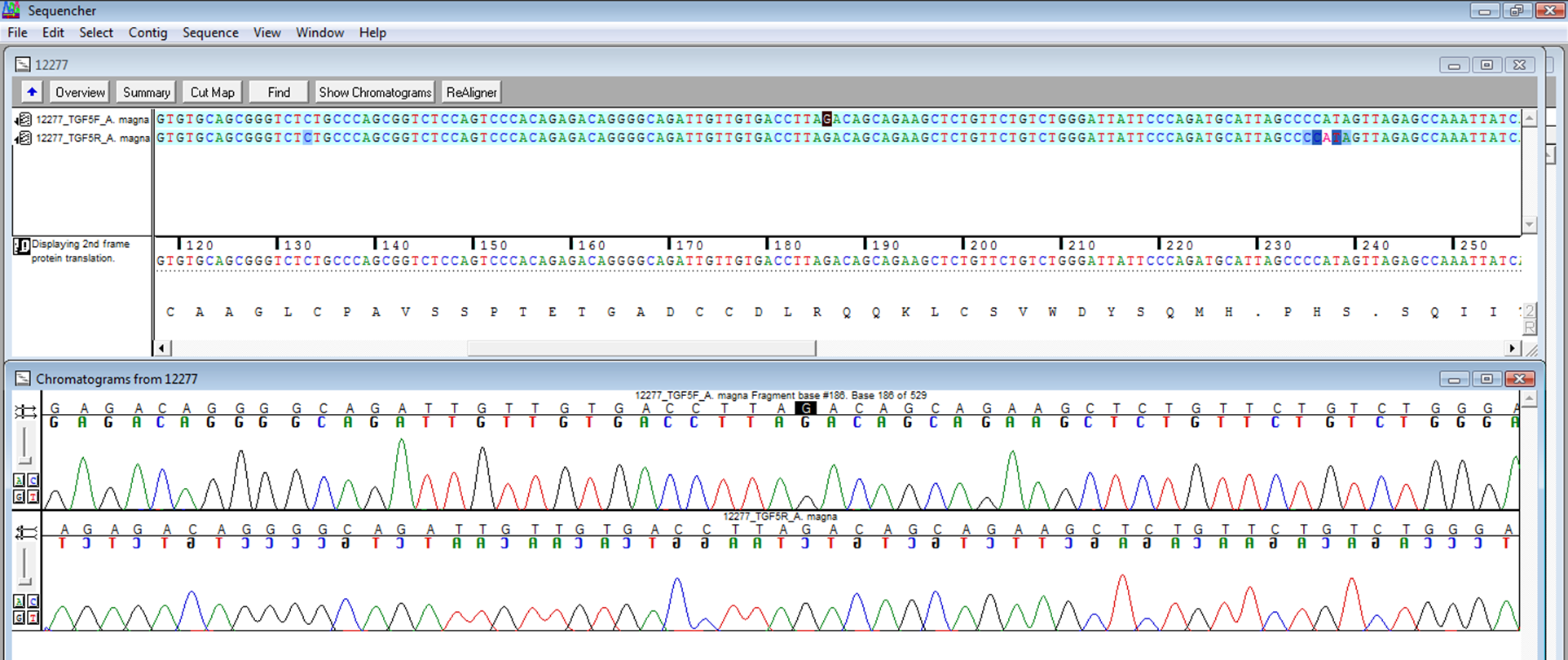
DNA sequences
Advantages: PCR-based
almost any tissue can be used
tiny amounts of tissue required
output is exact sequence of bases - can see lots of variation
Disadvantage
usually measures diversity within a single sequence
moderately expensive
Sequencing
Estimates of genetic variation: Haplotype diversity
e.g. mtDNA data
describes the numbers and frequencies of mtDNA haplotypes
Heterozygosity equivalent for haploid loci
Will often approach 1 if a high proportion if individuals have unique haplotypes
no assessment of the magnitude of the difference (just assesses whether they are different)
Nucleotide diversity (pi)
factors in both frequences and pairwise divergences of the different sequences
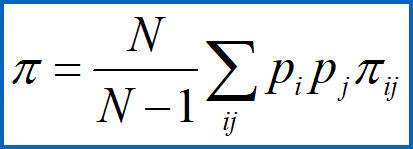
N = number of sequences in sample
pi and pj are frequency of sequences I and J in the sample and
piij is the proportion of sites that differ between sequences i and j