M1L7 R loops in health and disease
R-loops - nascent RNA formed during transcription can hybridise with DNA, leaving a ssDNA strand
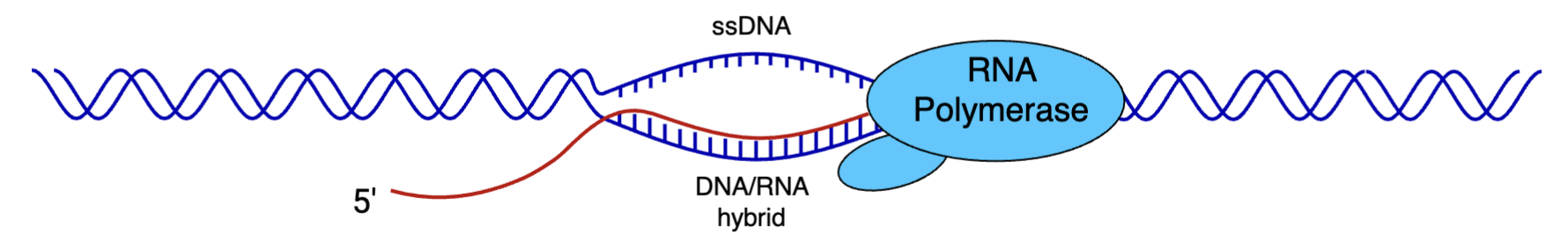
Formed in all living organisms
Preferentially formed on sequences enriched for GCs (as they form 3 hydrogen bonds, thus a thermodynamically more stable structure)
Occupy ~10% of the human genome
Important regulatory functions
Transcription - R-loops slow down RNA pol progression by reducing the processivity of the enzyme
DNA replication - Okazaki fragments form short, mini R loops
Epigenetics - regulate the formation of histone modifications and DNA methylation
Genome stability - ssDNA is unstable and can be subjected to SSB which can lead to DSBs
DNA damage response
Immune response - R loops can be processed, generating products that are exported to the cytoplasm where they bind with PRRs which can stimulate an immune response
CRISPR targeting - gRNA hybridising to target sequence in DNA, forming a trans R-loop which must compete with cis R-loops
Cis R loops: RNA transcribed from the same locus where it hybridises
Trans: RNA transcribed elsewhere and invades a different DNA locus
gRNA (trans) must compete with the nascent transcript (cis) for access to the DNA, if the cis R loop is stable it can block/interfere with Cas9 activity
Human disease - cancer and neurodegeneration
Molecular mechanisms to detect R-loops
S9.6 Ab - recognises RNA/DNA hybrids in non sequence-specific manner with high affinity
May have some background due to some binding to dsRNA, RNase H thus needed as a control (this should deplete the signal if it is specific)
DNA/RNA immunoprecipitation sequencing (DRIP-seq)/qPCR **

Lyse non-crosslinked cells, extract and lyse nuclei, sonicate to break RNA/DNA hybrids
Why non-crosslinked - RNA/DNA hybrids are thermodynamically more stable than DNA/DNA structures as it is a B fold whereas RNA/DNA is A fold (duplex is wider and bases are more stuck to each other, thus the hybrids can persist without cross linking), also to understand endogenous interactions rather than cross linking all RNA to DNA
IP with S9.6, wash and purify RNA/DNA hybrids
Can make a library or amplify using primers, if there is a lot of amplification that means there was a lot of binding/lot of hybrids in that region
Sequence/qPCR
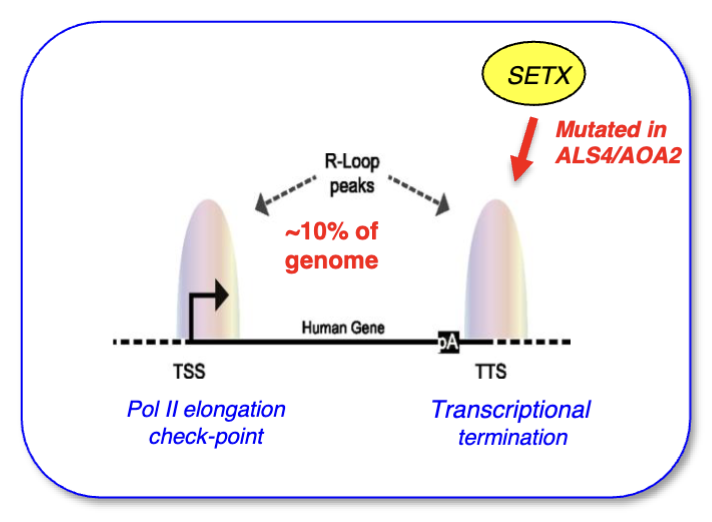
For all transcribed genes: major R-loop peak at TSS and TTS (latter signal is slightly smaller), and low (but not absent) signal throughout the gene
Peak at the promoter shifted slightly into the body of the gene - may be because you need to synthesise a bit of RNA first to start hybridising
Promoter R-loops important as polymerase checkpoint
R-loops at TTS push RNA pol backwards which slows down its progression and aids in termination, and then the hybrid needs to be opened to free the RNA
SETX helicase helps to separate the strand and allow exonuclease XRN2 to degrade a remaining fragment of RNA that was synthesised and is attached to RNA pol II downstream of the poly A site (torpedo model)
SETX is mutated in ALS/AOA2 (motor neuron disease)
Mass spectrometry to characterise R-loop proteome

Chromatin proteins, mRNA processing proteins, rRNA processing proteins,
Novel R loop binding factors - RNA binding proteins (R-loop turnover), nucleases (R-loop cleavage), helicases (R-loop resolution), DNA binding proteins (R-loop associated instability)
R-loop proteome
Topoisomerases eg. Top I - affects DNA compaction and R-loop expression
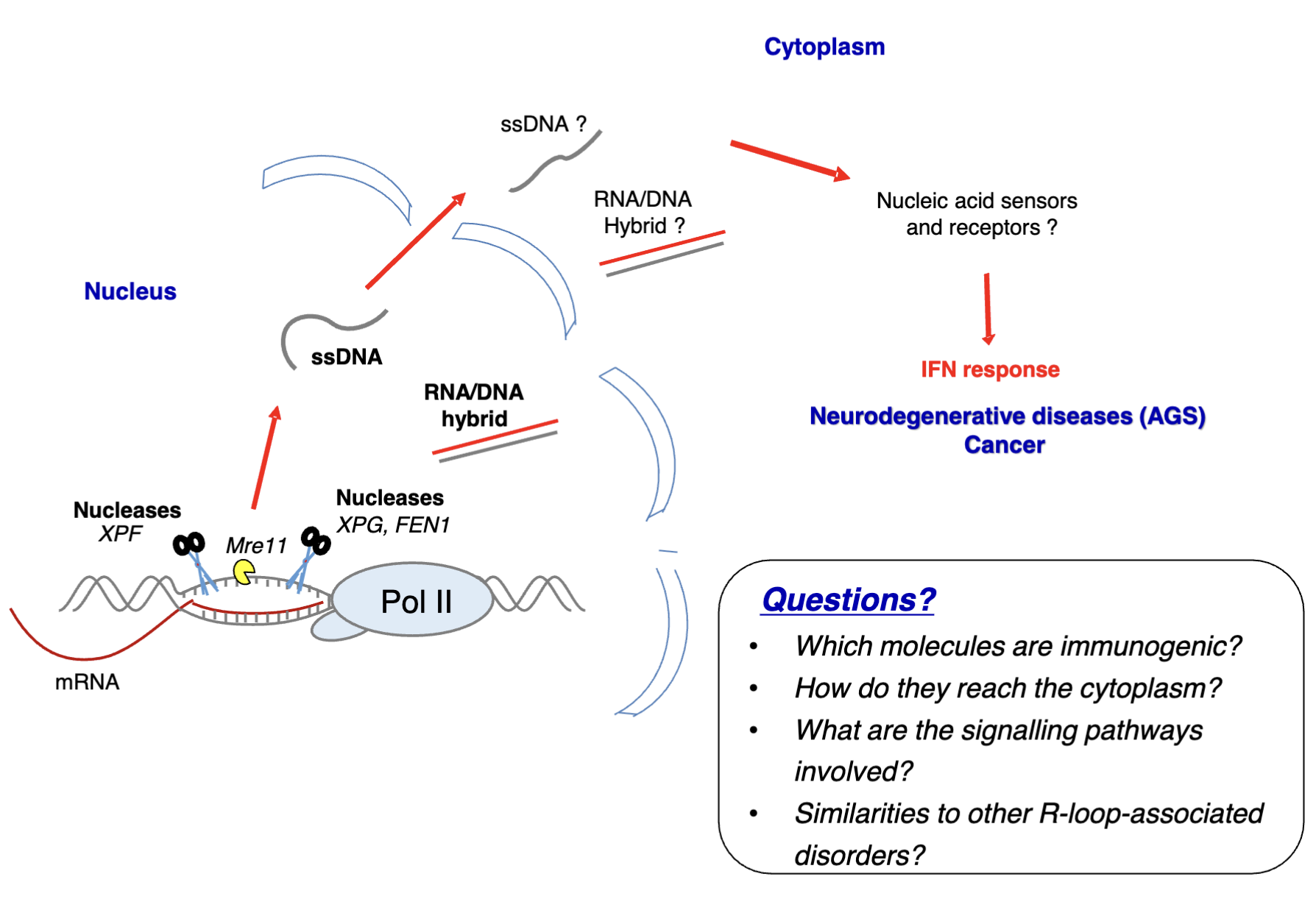
Nucleases eg. XPG/XPF endonucleases - involved in transcription-coupled NER and R-loop regulation if there is an extreme accumulation (eg. due to mutations in R loop regulators like APOBECs)
This generates DSBs which must be repaired, however this is preferable/a more controllable process to R-loop accumulation which can cause more damage
DNA fragments and DNA/RNA hybrids can be exported to the cytoplasm where they may be recognised by PRRs (eg. cGAS which acts as a DNA sensor) to generate inflammation
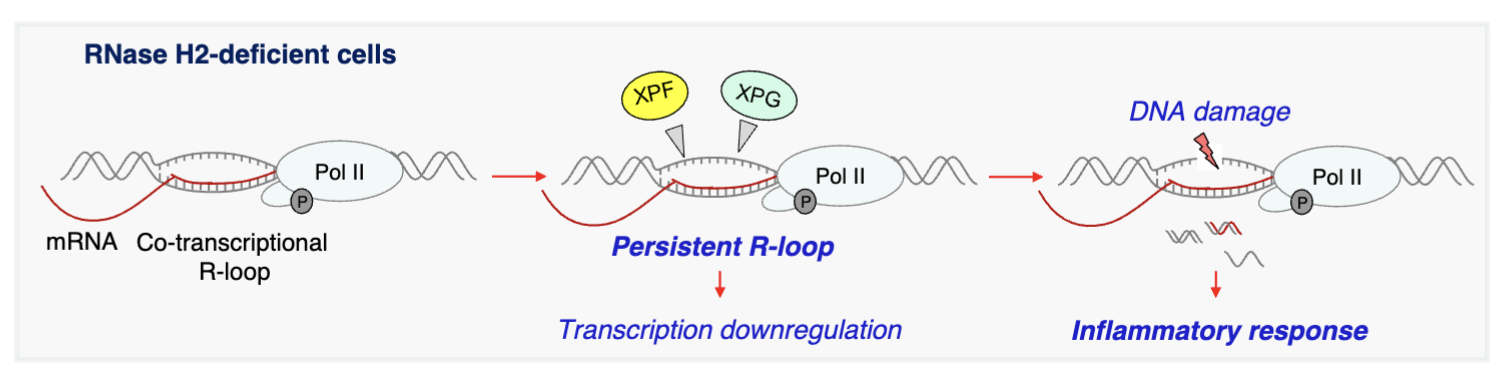
RNase H2 - degrades DNA/RNA hybrid, mutations in this cause neurodegenerative and inflammatory disease as they function to resolve R-loops to avoid DNA damage and inflammation
Helicases (eg. SETX, DHX9, AQR) - unwinding RNA/DNA hybrids (preferred pathway to degrading the hybrids), unclear whether each of the classes of helicases have specificity for certain genes, cell cycle stages, cell types, pathological conditions… or something else
m6A RNA modification machinery (eg. METTL3, YTDF1/2, hnRNPA2B1)
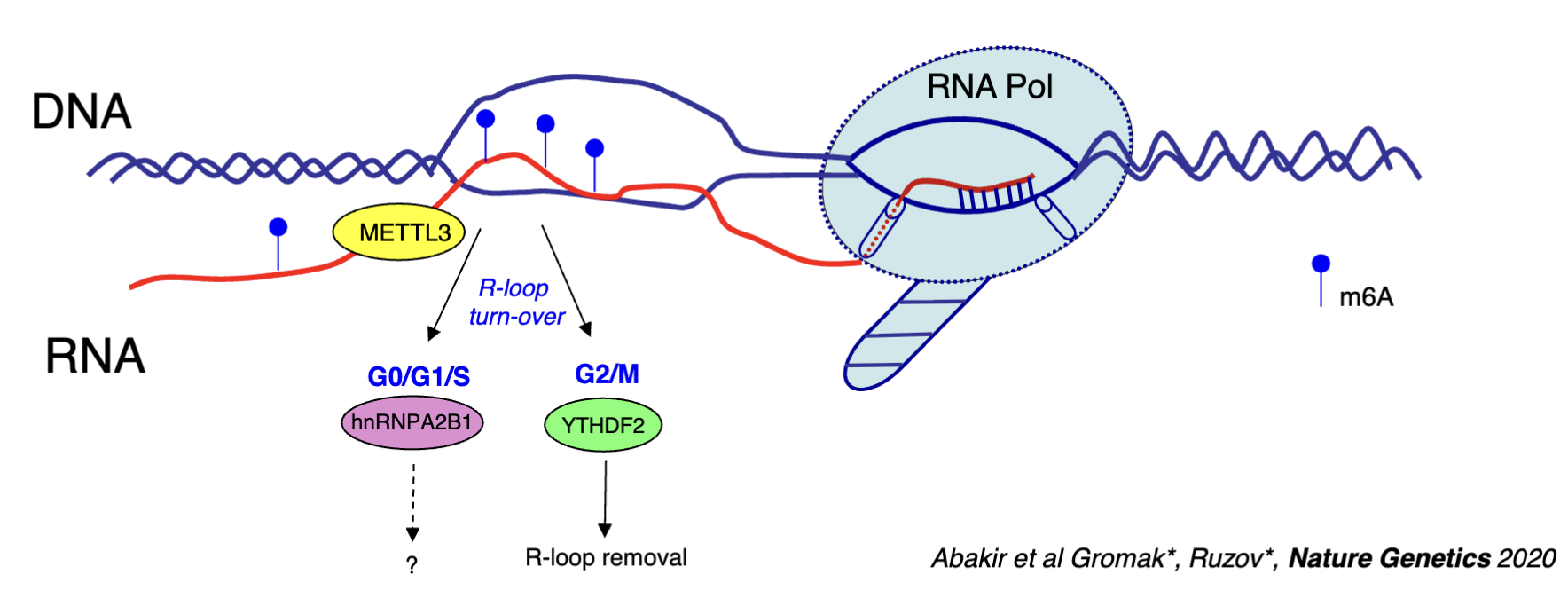
METTL3 travels with RNA pol II and acts co-transcriptionally to modify RNA (adds M6A modification which will be part of the hybrid)
M6A may increase the stability of the hybrid - some types of modifications in R loops could be implicated in disease pathology by increasing the stability and generating too many R loops
hnRNPA2B1 and YTHDF2 are readers of this modification
hnRNPA2B1 - recognises the R-loop in G0/G1/S phase, function unclear
YTHDF2 - R loop removal in G2/M phase
Deaminase (eg. APOBEC3B, AID)
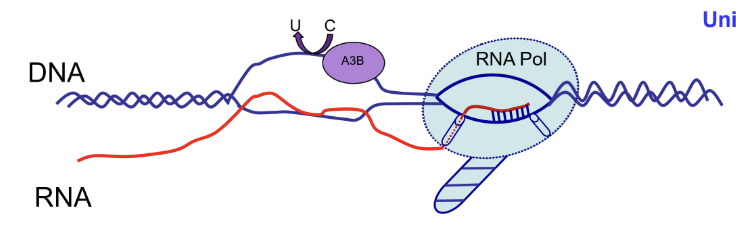
APOBEC regulates R loops and promotes cancer mutagenesis
APOBEC3B is a part of the APOBEC family of cytidine deaminases which converts C to U in ssDNA
This acts as an antiviral defense factor by editing viral retroelement DNA and introducing inactivating mutations
ssDNA generated in R loop formation is vulnerable to off-target APOBEC3B activity which can deaminate C into U, creating U:G mismatches
DNA breaks can also be caused
APOBEC3B is often overexpressed in tumours and causes clustered mutation (kataegis)
AID (homologue of APOBEC) has positive effect by doing class switching and creating antibody diversity in B cells
R-loops in disease
When pathological R-loops > physiological R-loops
Repeat expansion disease - R loops accumulate particularly in repetitive sequences, causing coding expansion (eg Huntington’s disease) or non-coding expansion (eg Friedreich’s ataxia)
Neurological disorders (ALS4/AOA2) - STX mutations
Alcardi-Goutieres Syndrome (AGS) - RNase H2 mutations
Cancer - dysregulation of transcription and promoting cancer mutagenesis
R loops in cancer
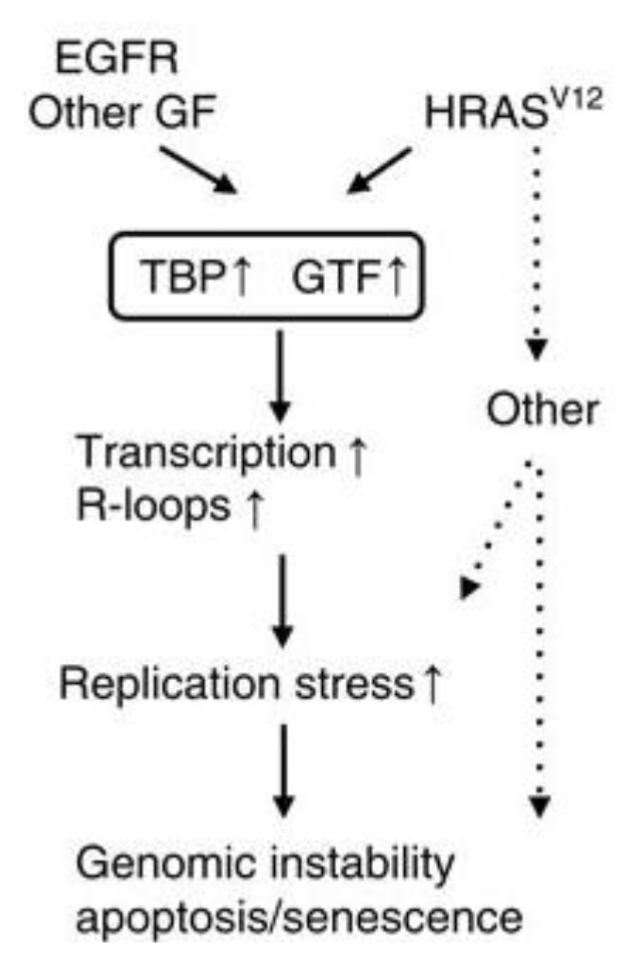
Oncogene activation (eg RAS or EGFR) increases global transcription and R loop accumulation which causes replication stress and genomic instability
EGFR and HRASV12 get overactivated in cancer which upregulates components of transcription machinery like TATA binding protein and general TFs which increase transcription initiation rate genome-wide
This causes RNA pol II to be overactive and more genes are transcribed simultaneously
Excessive transcription generates more opportunities for R loop formation which can stall replication forks during transcription-replication collisions and cause replication stress
This causes fork collapse and DNA DSBs which may cause chromosomal rearrangements, DNA copy number changes and mutation accumulation
Cells under extreme stress may undergo apoptosis or senescence