CLINICAL-PHARMACY-VENOUS-THROMBOEMBOLISM (3).docx
CLINICAL PHARMACY
VENOUS THROMBOEMBOLISM
I. DEFINITION. Venous thromboembolic disease (VTED) occurs when one or more of the elements of Virchow’s triad are present, resulting in deep venous thrombosis (DVT) and/or pulmonary embolism (PE).
• Vascular injury
• Venous stasis
• Hypercoagulable state (i.e., decreased protein C, protein S, or antithrombin)
II. INCIDENCE. It is estimated that the annual incidence of venous thromboembolism (VTE) events exceeds 600,000, and the number of VTE-associated deaths is 296,370 annually.
III. RISK FACTORS FOR VTE
A. Patient specific and those associated with medical illness and surgical procedures
1. Surgery
2. Trauma (major or lower extremity)
3. Immobility or paresis
4. Malignancy
5. Cancer therapy (hormonal, chemotherapy, or radiotherapy)
6. Previous VTE
7. Increasing age (! 40 years of age)
8. Pregnancy and the postpartum period
9. Estrogen-containing oral contraceptives or hormone-replacement therapy
10. Selective estrogen-receptor modulators
11. Acute medical illness
12. Heart or respiratory failure
13. Inflammatory bowel disease
14. Nephrotic syndrome
15. Myeloproliferative disorders (i.e., diseases in which malignant (cancer) bone marrow cells multiply and spread to the blood).
16. Paroxysmal nocturnal hemoglobinuria
17. Obesity
18. Smoking
19. Varicose veins
20. Central venous catheterization
21. Inherited or acquired thrombophilia (e.g., deficiency of antithrombin, protein C, or protein S; activated protein C resistance; antiphospholipid antibody; lupus anticoagulant)
IV. PREVENTION AND TREATMENT
A. Nonpharmacologic prevention. Mechanical methods of prophylaxis are recommended, primarily in patients who are at high risk of bleeding, and may include external pneumatic compression, graduated compression stockings, or venous foot pumps. These devices increase venous outflow and/or reduce stasis within the leg veins.
B. Pharmacologic prevention. VTED can be prevented by counteracting increased blood coagulability with unfractionated heparin (UFH) ; oral anticoagulant therapy with a vitamin K antagonist, such as warfarin; low-molecular-weight heparin (LMWH); a synthetic pentasaccharide; or the new oral agents (factor Xa inhibitor and factor IIa inhibitor.
V. PHARMACOLOGIC AGENTS. New recommendations for treatment of VTED have been promulgated that suggest a hierarchical approach to selection of pharmacologic agents for managing VTED. These recommendations are listed in Table 35-1 and each of the recommended pharmacologic agents is discussed in the following sections:
A. Unfractionated heparin
1. Indications. Patients with proven VTED may receive concomitant UFH for acute treatment and warfarin therapy acutely, followed by warfarin therapy for continued prevention of recurrence of VTED (Table 35.1), unless contraindications to warfarin (e.g., pregnancy) are present.
2. Mechanism of action. The major mechanism by which heparin blocks coagulation is by catalyzing the inhibition of thrombin. UFH acts as an anticoagulant by catalyzing the inactivation of thrombin (factor IIa), activated factor X (factor Xa), and activated factor IX (factor IXa) by antithrombin.
3. Pharmacokinetics. The mechanisms of heparin clearance are complex.
A. Heparin binds to a number of plasma proteins other than antithrombin, which competes with antithrombin heparin binding.
B. UFH is cleared by rapid-phase (cellular) elimination followed by a more gradual (renal) clearance, which can best be explained by a combination of saturable and nonsaturable first-order kinetic models.
C. When administered in fixed doses, the anticoagulant response to UFH varies among patients and within the same patient (i.e., interpatient and intrapatient variability). This variability is caused by differences in patients’ plasma concentrations of heparin-neutralizing proteins and rates of heparin clearance.
4. Administration and dosage
a. UFH’s therapeutic effect is hastened by administration of a loading dose, which may be empirically selected (e.g., 5000-units bolus given intravenously) or individualized by the patient’s dosing weight.
(1) The weight-based approach has resulted in the use of loading doses varying from 70 to 100 units/kg.
(2) In some instances, the indication for which heparin therapy is being initiated is to be considered, with 80 units/kg being used for all thrombotic indications other than suspected or proved PE, for which up to 100 units/kg may be used.
b. Variable approaches to continuous dosing have been employed.
(1)Empiric dosing of 1000 units/hr may be used but may result in subtherapeutic or supratherapeutic outcomes (i.e., an activated partial thromboplastin time [aPTT] below or above the targeted range).
(2) Another approach to continuous dosing includes commencing with a fixed dose (other than the empiric dose of 1000 units/hr). One such approach may see an initial loading dose followed by 32,000 units/24 hrs by continuous infusion.
(3) Yet another approach validated in the medical and pharmaceutical literature uses a weight-based dosing nomogram for commencing UFH therapy that varies between 15 and 25 units/kg/hr.
(a) Lower doses are used initially for most thrombotic indications other than PE.
(b) PE requires more aggressive therapy (i.e., up to 25 units/kg/hr) based on the consideration that the clearance of heparin may be increased, thus necessitating an increased dose.
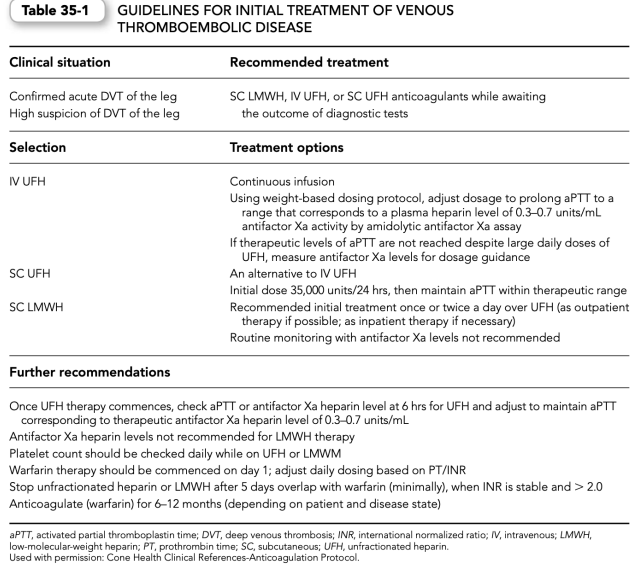
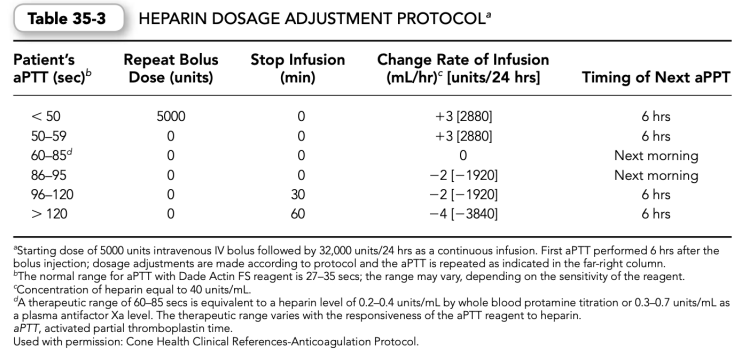
(4)Initial heparin weight-based dosing nomograms and subsequent adjustment proto- cols have been developed to assist the initial weight-based dosing efforts. Such protocols should be developed for a specific aPTT reagent (Tables 35-2 and 35-3).
5. Monitoring the effects of UFH. The anticoagulant effects of UFH are usually monitored by the aPTT. The aPTT should be obtained at baseline before commencing therapy and then monitored 6 hrs after commencing heparin therapy. Subsequent dosing adjustments are based on the results of this and additional aPTTs.
a. The aPTT ratio used to determine therapeutic effect is measured by dividing the observed aPTT by the mean of the normal laboratory control aPTT.
b. Traditionally, it was taught that for many aPTT reagents, a therapeutic effect was achieved with an aPTT ratio of 1.5 to 2.5.
c. However, because aPTT reagents may vary in their sensitivity, it is inappropriate to use the same aPTT ratio (i.e., 1.5 to 2.5) for all reagents. The therapeutic range for each aPTT reagent should be calibrated to be equivalent to a heparin level of 0.2 to 0.4 units/mL by whole blood (protamine titration) or to an antifactor Xa level (i.e., plasma heparin level) of 0.3 to 0.7 units/mL collected at the sixth hour for UFH.

B. Oral anticoagulants—warfarin
1. Indications
a. Warfarin is proven effective in the
(1) primary and secondary prevention of VTED;
(2) prevention of systemic arterial embolism in patients with tissue and mechanical prosthetic heart valves or atrial fibrillation;
(3) prevention of acute myocardial infarction (MI) in patients with peripheral arterial disease; and
(4) prevention of stroke, recurrent infarction, and death in patients with acute MI.
b. Warfarin may also be used in patients with valvular heart disease to prevent systemic arterial embolism, although its effectiveness has never been demonstrated by a randomized clinical trial.
Mechanism of action
a. Oral anticoagulants (e.g., warfarin) are vitamin K antagonists, producing their anticoagulant effect by interfering with the cyclic interconversion of vitamin K and its 2-, 3-epoxide (vitamin K epoxide).
b. Inhibition of this process leads to the depletion of vitamin KH2 and results in the production of hemostatically defective, vitamin K–dependent coagulant proteins or clotting factors (prothrombin or factors II, VII, IX, and X).
c. These vitamin K–dependent coagulant proteins or clotting factors (factors VII, IX, X, and II, respectively) decline over 6 to 96 hrs.
3. Pharmacokinetics
a. Warfarin is a racemic mixture of roughly equal amounts of two optically active isomers: the R and S forms.
b. Warfarin is rapidly absorbed from the gastrointestinal tract and reaches maximal blood concentrations in healthy volunteers in 90 mins. c. Dose response to warfarin is influenced by
(1) Pharmacokinetic factors (i.e., differences in absorption and metabolic clearance)
(2) Pharmacodynamic factors (i.e., differences in the hemostatic response to given concentrations of warfarin)
(3) Technical factors—for example, inaccuracies in prothrombin time (PT) and international normalized ratio (INR) testing and reporting
(4) Patient-specific factors—for example, diet (increased intake of green, leafy vegetables), poor patient compliance (missed doses, self-medication, alcohol consumption) and poor communication between patient and physician (undisclosed use of drugs that may interact with warfarin) (Table 35-4).

4. Administration and dosage
a. Warfarin, a coumarin compound, is the most widely used oral anticoagulant in North America. Although it is primarily administered orally, an injectable preparation is available in the United States.
b. Commence oral anticoagulant therapy in the inpatient setting with the anticipated daily maintenance dose of warfarin, which can be variable.
c. The initial dose of warfarin therapy can be flexible (Table 35-5).
(1) Patient-specific parameters used to determine the initial dose of warfarin include the patient’s weight (e.g., obesity, concurrent use of interacting drugs known to inhibit the anticoagulant effect of warfarin and the desired rapid anticoagulant effect).
(2) Based on these patient-specific parameters, some clinicians may use a larger initial dose of warfarin (e.g., 7.5 to 10 mg), which should not be misconstrued as a loading dose. For patients sufficiently healthy to be treated as outpatients—specific to VTE treatment—the suggestion is to commence warfarin therapy with 10 mg daily for the first 2 days, followed by dosing based upon the INR response.
d. The initial dose of warfarin should be overlapped with UFH, LMWH, or a pentasaccharide for a minimum of 5 days for treating established embolic disease to 7 days (Table 35-1).
e. The duration of warfarin therapy depends on each patient’s indication(s) for use (Table 35-6).
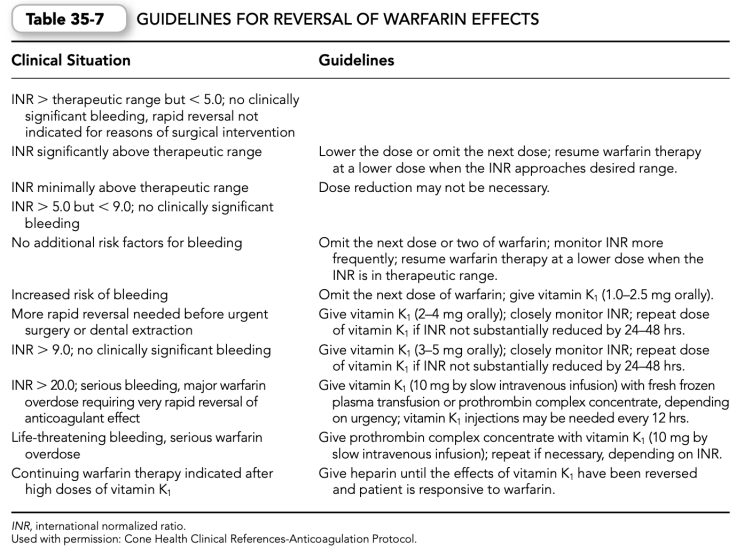
f. Reversal of warfarin effects may be necessary owing to an elevated INR or bleeding complications associated with oral anticoagulant therapy (Table 35-7).
Monitoring warfarin therapy. PT and INR monitoring are usually to be performed at baseline and daily when commencing warfarin in the inpatient acute care setting. Commencing warfarin in the outpatient setting after establishing a baseline INR may see the subsequent INR determinations being performed based upon well-established protocols specific to the outpatient setting. In either setting (inpatient or outpatient), INRs will be performed daily on commencing oral anticoagulant therapy (e.g., warfarin), until such time that the INR has been found to be therapeutic. a. Laboratory monitoring is performed by measuring the PT for calculation of the INR.
(1) The PT is responsive to suppression of three of the four vitamin K– dependent pro- coagulant clotting factors (prothrombin or factors II, VII, and X).
(2) The common commercial PT reagents vary markedly in their responsiveness to coumarin- induced reduction in clotting factors; therefore, PT results reported using different reagents are not interchangeable among laboratories.
b. The problem of variability in responsiveness of PT reagents has been overcome by the intro- duction of a standardized test known as the INR.
(1) The INR is equal to:
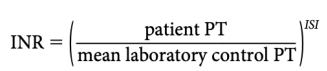
where ISI (international sensitivity index) is a measure of the responsiveness of a given thromboplastin to reduction of the vitamin K– dependent coagulation factors. The lower the ISI, the more responsive the reagent and the closer the derived INR will be to the observed PT ratio.
(2) Current recommendations suggest two levels of therapeutic intensity: a less-intense range corresponding to an INR of 2.0 to 3.0, and a more intense range corresponding to an INR of 2.5 to 3.5. The range corresponds to the indication (Table 35-8).
(3) Once the desired therapeutic INR has been achieved for 2 consecutive days (e.g., for concomitant heparin plus warfarin overlap therapy), follow-up INR monitoring can be per- formed according to the following protocol:
(a) Week 1: monitor INR two or three times
(b) Week 2: monitor INR two times
(c) Weeks 3 to 6: monitor INR once a week
(d) Weeks 7to14: monitor INR once every 2 weeks
(e) Week 15 to end of therapy: monitor INR once every 4 weeks (if INR dose responsiveness remains stable; if dose adjustment is necessary, a more frequent monitoring schedule is employed until stable dose responsiveness is achieved)
c. Upon commencing oral anticoagulant therapy, prolongation of the PT/INR does not occur until depletion of the vitamin K–dependent procoagulant clotting factors occurs. This delay is variable over 2 to 4 days. During this delay, if active venous thrombosis is present, either UFH, LMWH, or a synthetic pentasaccharide is concomitantly commenced to adequately anticoagulate the patient while awaiting the therapeutic effect of warfarin.

C. Low-molecular-weight heparin
1. Indications
a. LMWH indications vary by manufacturer.
b. Each of the LMWHs has been evaluated in a large number of randomized clinical trials and has been proven to be safe and efficacious for the prevention and treatment of VTE.
c. To date, different LMWHs have been evaluated for their role in
(1) prevention of venous thrombosis,
(2) treatment of VTED, and
(3) management of unstable angina pectoris/non–Q-wave MI.
2. Chemistry. LMWHs are fragments of standard commercial-grade heparin produced by either chemical or enzymatic depolymerization. LMWHs are approximately one-third the size of heparin. Like heparin, which has a mean molecular weight of 15,000 Da (range 3000 to 30,000 Da), LMWHs are heterogeneous in size with a mean molecular weight of 4000 to 5000 Da (range 1000 to 10,000 Da).
3. Mechanism of action
a. LMWHs achieve their major anticoagulant effect by binding to antithrombin through a unique pentasaccharide sequence that enhances the ability of antithrombin to inactivate factor IIa (thrombin) and factor Xa.
(1) Heparin and LMWHs catalyze the inactivation of factor IIa (thrombin) by binding to antithrombin through the unique pentasaccharide sequence and to thrombin to form a ternary complex. A minimum chain length of 18 saccharides (including the pentasaccharide sequence) is required for ternary complex formation.
(a) Virtually, all heparin molecules contain at least 18 saccharide units.
(b) Only 20% to 50% of the different LMWHs contain fragments with 18 or more saccharide units.
(c) Therefore, compared with heparin, which has an anti-factor Xa to antifactor IIa binding affinity ratio of approximately 1:1, the various commercial LMWHs have an antifactor Xa to antifactor IIa binding affinity ratio varying from 2:1 up to 4:1, depending on their molecular size distribution (Table 35-9).
(2) In contrast, inactivation of factor Xa by antithrombin does not require binding of the heparin molecules to the clotting enzyme. Therefore, inactivation of factor Xa is achieved by small molecular weight heparin fragments provided that they contain the high-affinity pentasaccharide.
b. The antithrombotic and hemorrhagic effects of heparin have been compared with LMWHs in a variety of experimental animal models.
(1) When compared on a gravimetric basis, LMWHs are said to cause decreased potential for hemorrhagic episodes.
(2) These differences in the relative antithrombotic to hemorrhagic ratios among these polysaccharides could be explained by the observation that LMWHs have less inhibitory effects on platelet function and vascular permeability.
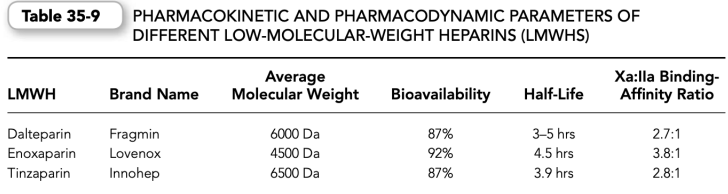
4. Pharmacokinetics. The plasma recoveries and pharmacokinetics of LMWHs differ from heparin because of differences in the binding properties of the two sulfated polysaccharides to plasma proteins and endothelial cells.
a. LMWHs bind much less avidly to heparin-binding proteins than heparin; a property that contributes to the superior bioavailability of LMWHs at low doses and their more predictable anticoagulation effect.
b. LMWHs do not bind to endothelial cells in culture; a property that could account for their longer plasma half-life and their dose-independent clearance. Principally, the renal route clears LMWHs; therefore, the biologic half-life of LMWHs is increased in patients with renal failure.
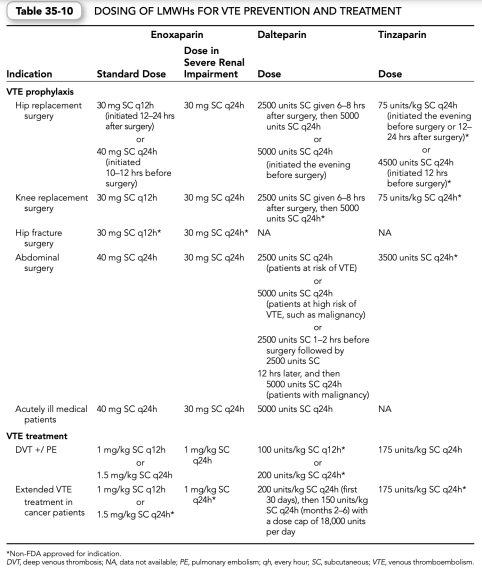
5. Administration and dosage
a. Dosing of LMWHs is disease state and product specific; different doses are administered based on the indication for use and the manufacturer of the specific LMWH. Table 35-10 shows manufacturer’s suggested, U.S. Food and Drug Administration (FDA) approved dosing for specific indications.
D. Synthetic pentasaccharide
1. Indications
a. Synthetic pentasaccharide (fondaparinux [Arixtra]) is indicated for
(1) thromboprophylaxis against DVT/PE after
(a) hip fracture surgery
(b) hip fracture surgery, extended prophylaxis
(c) knee replacement surgery
(d) hip replacement surgery
(e) abdominal surgery
(2) treatment of
(a) acute DVT
(b) acute PE
Chemistry. Synthetic pentasaccharide is a selective factor Xa inhibitor. The molecular weight of the synthetic pentasaccharide product is 1728 Da.
3. Mechanism of action
a. The antithrombotic activity of fondaparinux is the result of antithrombinmediated selective inhibition of factor Xa. Neutralization of factor Xa interrupts the blood coagulation cascade and thus inhibits thrombin formation and thrombus development.
b. Fondaparinux does not inactivate thrombin (activated factor II) and has no known effect on platelet function.
4. Pharmacokinetics
a. After subcutaneous administration, the drug is completely bioavailable, and steady-state peak plasma levels are achieved in approximately 3 hrs after administration of the dose.
b. The elimination half-life is 17 to 21 hrs, enabling once daily dosing. c. The drug does not seem to be metabolized, appears in the urine in active form, and is renally eliminated.
5. Administration and dosage
a. Fondaparinux must not be administered intramuscularly.
b. For prophylaxis against VTE, the drug should not be used in patients with body weight <50 kg because the incidence of major bleeding was found to double in this patient population during clinical trials.
c. For prophylaxis against VTE, a usual dose of 2.5 mg subcutaneously once daily for 5 to 9 days is recommended for all prophylaxis indications. The initial dose should be started 6 to 8 hrs after surgery when hemostasis is established.
d. For treatment of established VTE, administer a once daily subcutaneous dose as follows:
(1) Patients with body weight between 50 and 100 kg: 7.5 mg
(2) Patients with body weight < 50 kg: 5 mg
(3) Patients with body weight >100 kg: 10 mg
6. Cautions
a. Contraindications
(1) Severe renal impairment
(2) Patients weighing " 50 kg when used for prophylaxis
(3) Patients with active major bleeding
(4) Bacterial endocarditis
(5) Thrombocytopenia associated with positive in vitro test for antiplatelet antibody in the presence of fondaparinux
(6) Known hypersensitivity to fondaparinux
b. Precautions
(1) Conditions or procedures that may enhance the risk of severe bleeding (e.g., trauma, hemophilia, gastrointestinal ulceration, concurrent use of antiplatelet agents, history of cerebrovascular hemorrhage, severe uncontrolled hypertension)
(2) Renal impairment
(3) Heparin-induced thrombocytopenia
(4) Neuraxial anesthesia and indwelling epidural catheter use
(5) Elderly patients
(6) Pregnancy category B and lactating (the drug is excreted into breast milk)
(7) Protamine is ineffective as an antidote
E. Oral factor Xa inhibitor
Indications
Direct factor Xa inhibitor (rivaroxaban [Xarelto])
Thromboprophylaxis against DVT/PE after
(a) elective hip replacement surgery
(b) elective knee replacement surgery
To reduce the risk of stroke and systemic embolism in patients with atrial fibrillation.
Chemistry. Direct competitive specific inhibitor of factor Xa
Mechanism of action
a. The antithrombotic activity of rivaroxaban is the result of direct specific inhibition of factor Xa.
b. Rivaroxaban potentially inhibits both free and clot-associated factor Xa activity and has no direct effect upon platelet aggregation.
4. Pharmacokinetics
a. After oral administration, the drug is approximately 80% bioavailable and is rapidly absorbed with an onset of action achieved in approximately 2 to 4 hrs after administration of the dose.
b The drug undergoes oxidative metabolism (mainly via CYP3A4) and hydrolysis and is eliminated one-third by renal excretion and two thirds by hepatic metabolism.
c. The elimination half-life is 5 to 9 hrs, enabling once daily dosing for the orthopedic prophylaxis indication.
5. Administration and dosage
A. For prevention of VTE in adult patients undergoing elective hip or knee replacement surgery, the dosage recommendation is 10 mg orally once daily. For elective hip replacement surgery, the duration of therapy should be 35 days. For elective knee replacement surgery, the duration of therapy should be 12 days minimally.
B. No routine monitoring of INR or aPTT is necessary for this drug.
6. Cautions
a. Contraindications
(1) Hypersensitivity to the active substance or to any of the excipients; clinically significant active bleeding; hepatic disease associated with coagulopathy and clinically relevant bleeding risk, pregnancy, and lactation.
b. Precautions
(1) Treatment with rivaroxaban is not recommended in the following patients: concomitantly treated systemically with strong concurrent CYP3A4 inhibitor and plasma glycoprotein (P-gp) inhibitors, i.e., azole antimycotics (such as ketoconazole, itraconazole, voriconazole, and posaconazole) or HIV protease inhibitors (e.g., ritonavir); with severe renal impairment (creatinine clearance [Clcr] <15 mL/min); and, due to lack of data, younger than 18 years of age; undergoing hip fracture surgery.
(2) Strong CYP3A4 inducers (e.g., rifampicin, phenytoin, carbamazepine, phenobarbital, or St. John’s Wort) should be used with caution because they may lead to reduced rivaroxaban plasma concentrations and thus may reduce efficacy.
(3) Special care is to be taken when neuraxial anesthesia or spinal/epidural puncture is employed.
F. Oral direct thrombin inhibitor
1.Indications
a. Direct factor IIa (thrombin) inhibitor (dabigatran [Pradaxa])(1) Reduce the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation.
2.Chemistry. The drug binds rapidly and specifically to both free and clot-bound thrombin whose inhibition results in prevention of thrombus.
3.Mechanism of action
a. The antithrombotic activity of dabigatran is the result of directly and selectively inhibiting factor IIa (thrombin).
4.Pharmacokinetics
A. After oral administration, the drug is rapidly absorbed, converted to active drug and eliminated primarily by renal excretion, with an onset of action achieved in approximately 1 hr after administration of the dose.
B. The drug undergoes conjugation (esterase catalyzed hydrolysis in liver or plasma).
C. The elimination half-life is 12 to 17 hrs, requiring twice daily administration for the atrial fibrillation indication.
5. Administration and dosage
a. For reduction of risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillations, the recommendation is 150 mg orally twice daily if renal function is greater than 30 mL/min Clcr. For renal function 15 to 30 mL/ min, the recommended dose is 75 mg orally twice daily. For renal function < 15 mL/min, the drug is not recommended. For patients with moderate renal impairment (Clcr 30–50 mL/min) consider reducing the dose of dabigatran to 75 mg orally twice daily if co-administered with dronedarone or systemic ketoconazole.
b. Routine monitoring (INR or aPTT) is not necessary for this drug.
6. Cautions
a. Contraindications
(1) Hypersensitivity to the active substance or to any of the excipients and clinically significant active pathologic bleeding.
b. Precautions
(1) Treatment with dabigatran increases the risk of bleeding and can sometimes cause significant and sometimes fatal bleeding. Risk factors for bleeding include certain medications known, to increase this risk (e.g., antiplatelet agents, heparin, fibrinolytic therapy, chronic use of nonsteroidal anti-inflammatory drugs (NSAIDs), and labor and delivery).
(2) The concomitant use of dabigatran with P-gp inducers (e.g., rifampin) reduces dabigatran exposure and should generally be avoided; P-gp inhibitors (e.g., ketoconazole, verapamil, amiodarone, and clarithromycin) do not require dose adjustment. These results should not be extrapolated to other P-gp inhibitors.
(3) Special care is to be taken when neuraxial anesthesia or spinal/epidural puncture is employed.