Lab F
Introduction:
Two gels will be used
Will wants to figure out the size and number of individuals subunits of a given protein using the SDS-PAGE technique
We will also determine the quality and the quantity of the PCR product we made use gel electrophoresis and the spectrophotometry
Part A:
Proteins have different structures
Gel Filtration: provides an estimate of the molecular wieght of native state proteins
SDS with a reducing agent separates protein subunit and separate through a polyacrylamide gel electrophoresis
How does the SDS-Page technique work?
referred to as denaturing electrophoresis
molecular weight is estimated comparing the migration of a denatured protein/unfolded protein/
SDS → anionic detergent sodium dodecyl sulfate
Is mixed with a reducing agent to create a loading buffer meant to break down disulfide bonds
SDS disrupts noncovalent interactions between subunits of proteins which causes protein unfolding
proteins will become negatively charged with SDS
proteins are added to the gel the same as DNA and will run from negative to positive based on their size but will use a polacrylamide gel instead
Acqua Stain will be used to stain the proteins in order to visual their runs
Part A Procedure:
get a protein samples from the TA
denature them in the heat block and then load it into a well
run gel at 250 V for 25 minutes and then stain with acquastain
will paste the graph on the worksheet
will use the curve to determine the size of an unknown polypeptide
(confused)
Part B: Agarose Gel Electrophoresis and Spectrophotometry
Electrophoresis: provides a qualitative overview of DNA purity
allow us to know whether amplified DNA is the correct size
agarose gel will be used here instead
Gels will be stained with gel green which will intercalate between base pairs and will be visualized under a blue LED lights
Spectrophotometry: provide a quantitative measure of the DNA
absoroption of 260/280 indicated good nucleic acid purity
to estimate the concentration of DNA, multiply the concentration of the sample by 50
we will be determine the amount of DNA in the PCR product and will use the ratio to determine the nucleic acid purity
Part B Procedure:
get a tube and mix the pertaining liquids and flick it, load into the gel
lowkey was confused there watch the video for help
Spectrophotometry to check DNA concentration
make a cuvet and then mixe with a pipette and read it with the spectrophotometer taking the 260 and 280 readings
warnings:
some irritations from gels and buffeds
gel green can also cause irritation
TBE buffer as well
everything goes in the regular trash
Video 1:
questions:
When is Text 2 for the Human Physiology lab due
Friday of Week 6 at NOON
The protein materials in LS7L (Instruments & Materials) are identified by the color 'orange'.
False
DNA = Orange
Proteins = Green
When loading a gel, why is it important to only push to the first stop of the pushbutton on the pipetter?
Pushing to the second stop may cause bubbles to blow your sample out of the gel well.
According to the lab safety sheet, which of the following is a potential hazard you will face in the Agarose and Polyacrylamide Gels lab?
Chemicals causing respiratory tract, eye and skin irritation
You will be loading two different types of gels. Which gel setup is shown here and what are we using it for?
PAGE / Protein (standing up thing that is labeled green)
Remember the Polyacrylamide gel will be used with the protein
This is the other type of gel, the agarose gel. What do we use it for in lab this week?
PCR product size
Agarose gel will be used with the Gel electrophoresis one
DNA will move through the gel towards the positive electrode because...
DNA is naturally negatively charged and the negative charge is attracted to the positive electrode.
we will not be treating the DNA with SDS, that is inly for the protein
looking at both quantitative and qualitative success of PCR
you will start by loading the protein sample where the TA will start running the DNA
you will get the DNA from the 96 well from last week and then loading it in the DNA gel
will analyze a mark protein
Part 2 of the Lecture Videos:
questions:
You add 5ul of your PCR product solution into 495 ul water. What is the dilution factor?
100
we are supposed to dilute our DNA once we collect the sample from the well
If the ratio of OD260/OD280 is 1.5 the DNA prep is pure
False should be around 1.8
The concentration of the DNA can be determined by using the OD280 reading.
False, it should be the 260 reading
You read an Absorbance of 1.00 at 260nm & 0.56 at 280nm. The DNA sample is…..
Pure
You read an Absorbance of 1.00 at 260nm & 0.56 at 280nm. What is the DNA concentration in the cuvette?
50ug/ml (microgram/mililiters) cause this will be equal to 0.05 milligram/militers
The DNA concentration in the cuvette is 50ug/ml. When you prepared your DNA sample for the spec reading you used 5ul from your PCR reaction and added 495ul water. What is the DNA concentration in the PCR reaction?
5mg/ml
What is the size (bp) of the Extended Hypervariable Segment I (HVSI)?
651 BP
What size (bp) do we expect the PCR product to be?
549 = 16520 - 15972 + 1
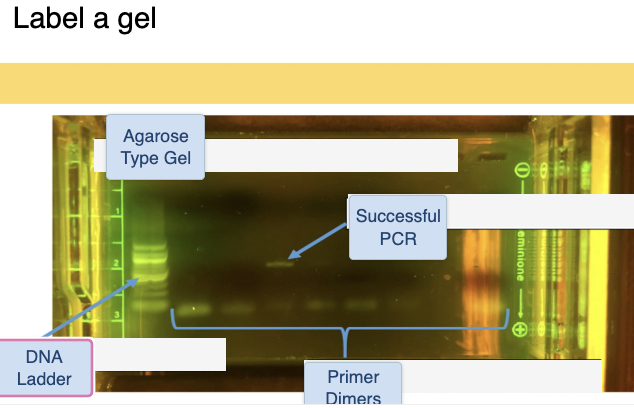
How to determine the purity of DNA prep
Determine concetration of DNA Prep
how to determine molecule length such as PCR product length
once we dilute we will take the reading on the spectrophotometry
reading done at 260 and 280
ratio about 1.8 good job isolating the DNA
less than 1.8 then protein is involved
The ratio between 260/280 of the DNA will determine the purity of the DNA prep
you will get the ratio once you have diluted the DNA with 495 of water and you take the reading
DNA concentration in the cuvette is determined by multiplying the 260 reading by 0.05
dna should equal 500 micrograms or 5 miligrams since the dilation factors was 100 so i got this from multiplying 50 and 0.05 by 100
molecule length: 651 base pairs
highest # - lowest # + 1 ( will give you the number of bp in a region
a success PCR will have the product at around 549 in all lanes with faint primer dimers
no bands and only once successful PCR means a failed PCR
might have failed due to having no DNA present?
part 3.1 (proteins)
questions
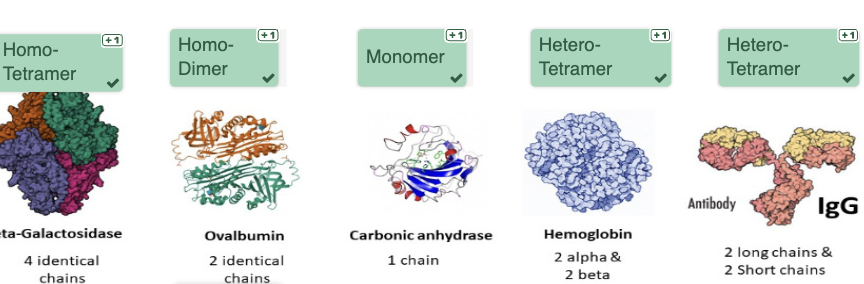
A protein dimer is an example of the
quarternary-structure of a protein. An Amino-Acid-Sequence is an example of the primary-structure. An alpha-helix is an example of a secondary-structure and a protein-domain is an example of the tertiary-structure.
Which if these molecular interactions is a covalent bond?
Disulfide bond
SDS (Sodium-dodecyl-sulfate) breakes cystein-linkages
False, the BME does this
Which type of protein gel will run proteins while keeping their enzymatic function intact?
Native Gel - Just buffer
In Size Exclusion chromatography larger molecules come out later than smaller molecules
False
Protein size is measured in
kilodaltons, sicne theyre so large
We have different molecular weight standards
homo-tetramer
will have the same subunits (4)
homodimer
will have two of the same subunits
Monomer
will have a single subunit
protein structure:
primary: single piece of protein
secondary: local interactions between amino acids
Tertiary: interactions between domains
quaternary: interactions between subunits
different types of bonds in protein structure
ionic: salt bridges?
hydrogen bonding between side chaings
dissulfide, covalent, and usually dont unfold
SDS and heat will be used to unfold a protein without cysteine bridges and will make a single unit
BME will work the same way but for proteins that have cysteine bridges
estimating a protein mass
there are different types of gel used to do this
native gel: keeps the protein intact
SDS: unfolds protein without cysteine bridges
Reducing Gel: proteins with cysteine bridges will unfold
proteins measured in kilodaltons
examples of protein purification
ultracentrifugation
separation based on size, shape, density
column chromatography
protein is added to a solvent with a lot of buffer which will dilute and separate?
separation by size
large come out faster
smaller molecules get trapped and come out slower
Choose the correct statement
My unknown protein can have one or more subunits
you can monitor each step of purification through the gel electrophoresis
lecture video 3.2
questions
In lab, protein will move through the gel towards the positive electrode because...
The protein is treated with SDS, which creates a uniformly negative charge in the molecule that is attracted to the positive electrode.
Choose the correct statement about your lab activity in the gels lab
You will be given a protein of known molecular weight (predetermined by gel filtration on the native molecule) but unknown subunit composition.
For this lab, we will use what type of stain for the PAGE gel?
acquastain
The loading dye's function is
to allow you to visualize how far the samples have run on the gel
Which of these functions shown in the image has the best fit for the data?
Exponential, since its the curve that fits most of the data points
An SDS-PAGE reducing gel
has SDS & beta-mercapto-ethanol and will break down disulfide bonds
You run a reducing SDS-PAGE of a protein with a known MW of 260kD. You get three bands (sizes 30kD, 40kD & 50kD). What is the protein composition?
3 of 50kD - 2 of 40kD - 1 of 30kD
will need to add up to 260 K
You run a non-reducing SDS-PAGE of the same protein. You get two bands. Why do you get only two bands?
Cysteine bridges between two 40kD and one 30kD Subunit
protein size estimation
PAGE gel setup
aqua stain will stain the protein in the gel after 15 minutes when added
you will measure the bands and create a curve out of them
we will have standards to compare are unknown to
We will first measure the know, and then the unknown, make the curve to get the protein estimate
Molecular weight has been determined by running the protein through the native gel
reducing gel will have BME
lecture video 3.3
questions
You need to separate very large proteins (>200kD) using a native Polyacrylamide Gel Electrophoresis (PAGE). Which percentage of polyacrylamide would you choose for optimal separation?
5 %
Why is it necessary to run standards alongside your samples every time in a PAGE gel?
To account for variations in gel and running conditions.
Choose the correct one
In LS 7L we use GelGreen to visualize the DNA, because it is a safe alternative to ethidium-bromide which is toxic
will have gel parameters
voltage
change the percentage of gel
how long the gel is ran for
gel will run depending on condition of parameters
with a higher percentage of the gel, the larger molecules will get stuck and not travel far
the higher the voltage the faster the protein will run
it could cook the gel
better to run with a lower voltage
time, running too early is bad when making the curve
running for too long then the bands will fall off the gel
gel is stained after its ran in the PAGE
in the agarose gel then stain is already there
we will used acquastain with the protein
we will use gel green with the DNA
the horizontal setup
Agarose / DNA / PCR Product Size / GelGreen
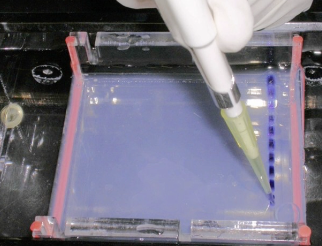
the vertical set up

PAGE / Protein / Protein Composition / AcquaStain