The Mitochondrial Genome
Learning Objectives
Describe features of the mitochondrial genome and how it differs from the nuclear genome
Explain the process of mitochondrial DNA replication
Describe features and causes of mitochondrial diseases
Describe diagnostic signs and tests for mitochondrial diseases
Describe the genetics of mitochondrial disease, including inheritance patterns and the phenomenon of heteroplasmy
Describe the process of mitochondrial replacement therapy
Features of Mitochondrial Genome
Mitochondria is important for ATP synthesis and is the energy currency of the cell
The energy is stored in nutrients and is broken down and converted into ATP through oxidative phosphorylation
Double-stranded circular molecule (16.6kb) which is 150,000 times smaller than chromosome 1:
Heavy (rich in G) and Light strand (less in G)
Multicopy genome
37 genes:
13 oxidative phosphorylation protein subunits
22 transfer RNAs - assembling proteins within mitochondria
2 ribosomal RNAs - mitochondrial protein synthesis
No introns - streamlined instruction manual with minimal interruptions
D-loop is a non-coding region where replication and transcription are initiated
Maternally inherited, no recombination (retains mutations - allows population geneticists to trace human migrations through haplogroups), you don’t inherit it from your father.
The mitochondrial genome encodes 13 proteins for OXPHOS. OXPHOS consists of 5 enzyme complexes in the inner membrane of mitochondria
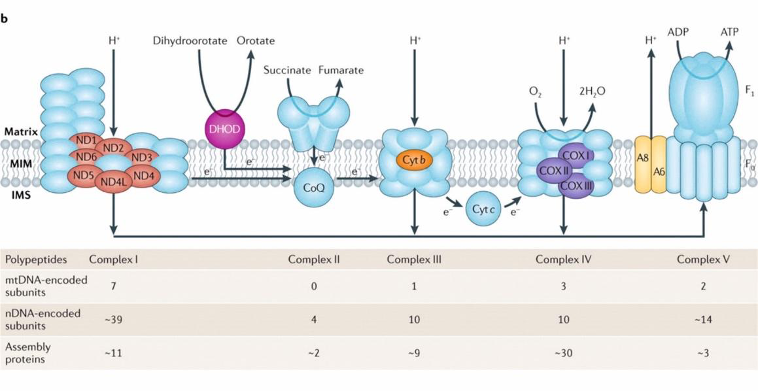
The first four complexes are the respiratory chain (RC) complexes (CI-CIV) and CV is the ATP synthase enzyme.
Three of the RC complexes pump protons across the membrane generating electrochemical potential across the membrane.
This potential is then utilized by CV to produce ATP.
This is known as chemiosmosis and was proposed by Peter Mitchell in the 1960s for which he won a Nobel Prize.
Non-coding region: contains regulatory sequences for replication and transcription
mtDNA replication starts in the origin of heavy strand
Exception to the Universal Genetic Code
• mtDNA uses a slightly altered genetic code:
• AUA codes for methionine (instead of isoleucine).
• UGA codes for tryptophan (instead of signaling “stop”).
• AGA and AGG are stop codons (not arginine).
• Think of mtDNA as using regional dialects in its language of life.
Endosymbiotic Theory
• Mitochondria evolved from ancient bacteria ingested by primitive cells. Over time, most of the bacterial DNA moved to the nucleus, leaving mtDNA as a remnant.

Mitochondrial DNA Replication
To make the 13 OXPHOS proteins mtDNA must be replicated, transcribed and translated. The proteins involved in these processes are encoded by nuclear genes and imported into mitochondria.
The mtDNA replication machinery needs:
1. Polymerase Gamma (POLG): The DNA Synthesizer
This enzyme is the central player in mtDNA replication.
• Structure:
Heterotrimer: Composed of three parts—one POLgA (the worker) and two POLgB (the helpers).
POLgA (Catalytic Subunit):
3’-5’ Exonuclease Domain: Acts as a “proofreader,” correcting mistakes during DNA synthesis. Imagine a carpenter checking for crooked nails and pulling them out.
Synthesizes the DNA strand using nucleotides.
POLgB (Accessory Subunits):
Enhances interactions with the DNA template.
Increases the activity and “processivity” (ability to keep synthesizing without stopping) of POLgA.
2. TWINKLE: The DNA Unzipper
TWINKLE is the helicase in the team, responsible for unwinding the double-stranded mtDNA so it can be replicated. Think of it as a zipper being pulled apart to reveal each strand.
Structure:
A hexamer, made of six identical TWINKLE subunits.
These subunits form a ring-like structure, encircling one strand of DNA while pushing the other strand aside.
Function:
Creates a single-stranded DNA template for POLG to work on.
Works like a motorized unwinder at the start of the DNA replication line.
3. Single-Stranded Binding Protein (SSBP): The DNA Stabilizer
Once TWINKLE separates the strands, SSBP steps in to ensure the single strands remain stable and ready for replication.
Functions:
Prevents Re-annealing: Stops the two strands from snapping back together, like keeping Velcro strips apart.
Protects Against Nucleases: Shields the exposed single-stranded DNA from enzymes that could degrade it.
Prevents Secondary Structures: Keeps the single strand from folding onto itself.
Stimulates TWINKLE Activity: Makes TWINKLE more efficient, ensuring smooth unwinding.
4. TFAM: The DNA Packager and Protector
Once replication is complete, the newly synthesized DNA needs protection and organization. That’s where Transcription Factor A Mitochondrial (TFAM) comes in.
Functions:
Binds to mtDNA and packages it into compact structures called nucleoids.
Acts like a histone (proteins that package nuclear DNA), helping mtDNA fit snugly in the mitochondrial matrix.
Shields mtDNA from damage, much like bubble wrap protects fragile items during shipping.
1. Unzipping: TWINKLE unwinds the double-stranded mtDNA, creating two single strands.
2. Stabilizing: SSBP binds to the separated strands to keep them stable and accessible.
3. Synthesis: POLG synthesizes a new complementary strand on each single-stranded template, with POLgB enhancing its performance.
4. Packaging: TFAM packages and protects the newly replicated mtDNA.
How They Work Together:
• TWINKLE clears the path by unwinding the DNA.
• SSBP holds the strands steady so POLG can do its job.
• POLG builds the new strands, while TFAM organizes and protects the completed work.
Replication Process:
Initiation
1. D-Loop Region:
This region is the control centre of mtDNA replication.
It contains the Origin of Heavy Strand (OH), where replication of the heavy strand starts.
Think of the D-loop as the starting gate for a relay race. The heavy strand begins the race, and the light strand waits for its turn.
2. RNA Primers:
To start DNA replication, RNA primers are synthesized by mitochondrial RNA polymerase (POLRMT). These short RNA sequences act as the “anchor” that DNA polymerase gamma (POLG) uses to attach and begin building new DNA strands.
Imagine RNA primers as the foundation for building a house. Without a solid base, construction can’t start.
Strand Displacement
1. Heavy Strand Replication:
Unwinding: The enzyme TWINKLE helicase unwinds the double-stranded DNA at OH, exposing the heavy strand as the template.
Replication: POLG begins synthesizing a new complementary strand using the heavy strand as a template. This occurs in the 5′ to 3′ direction.
As the heavy strand is replicated, the original heavy strand is displaced and stabilized by Single-Stranded Binding Proteins (SSBP) to prevent it from re-annealing or forming secondary structures.
2. Staggered Light Strand Replication:
After the heavy strand is about two-thirds replicated, the replication machinery encounters the Origin of Light Strand (OL).
At this point:
A stem-loop structure is formed at OL, which provides a signal for the light strand to begin its own replication.
POLRMT synthesizes an RNA primer at the OL to initiate replication of the light strand.
Think of this as a relay baton handoff: the light strand doesn’t start until the heavy strand has a significant head start.
Completion
1. Replication Finalization:
The heavy strand and light strand are synthesized until the entire circular mtDNA molecule is replicated.
Any remaining RNA primers are removed, and the gaps are filled in with DNA.
Segregation and Packaging:
Once both strands are replicated, they are separated into two daughter molecules.
Each daughter molecule is packaged into nucleoid structures by TFAM. These nucleoids protect and organize the mtDNA within the mitochondria.
Imagine TFAM as the librarian storing the freshly copied books (mtDNA) neatly into shelves (nucleoids).
• Initiation:
• Imagine mtDNA as a circular racetrack with two runners: the heavy strand and light strand.
• The heavy strand gets the starting whistle at OH and begins running, with RNA primers acting as their running shoes.
• Strand Displacement:
• As the heavy strand runner progresses, the light strand waits at OL for its cue. This staggered start ensures that they don’t interfere with each other on the track.
• Completion:
• Both runners complete their laps and hand off their replicated DNA to TFAM, which stores them in a safe locker.
Mitochondrial Disease
Rare monogenic disease
Most common: Oxidative Phosphorylation disorders - affect highly metabolic organs (heart, CNS and muscles) which are abundant in mitochondria
Can either affect one organ or several organ systems
Clinical Features:
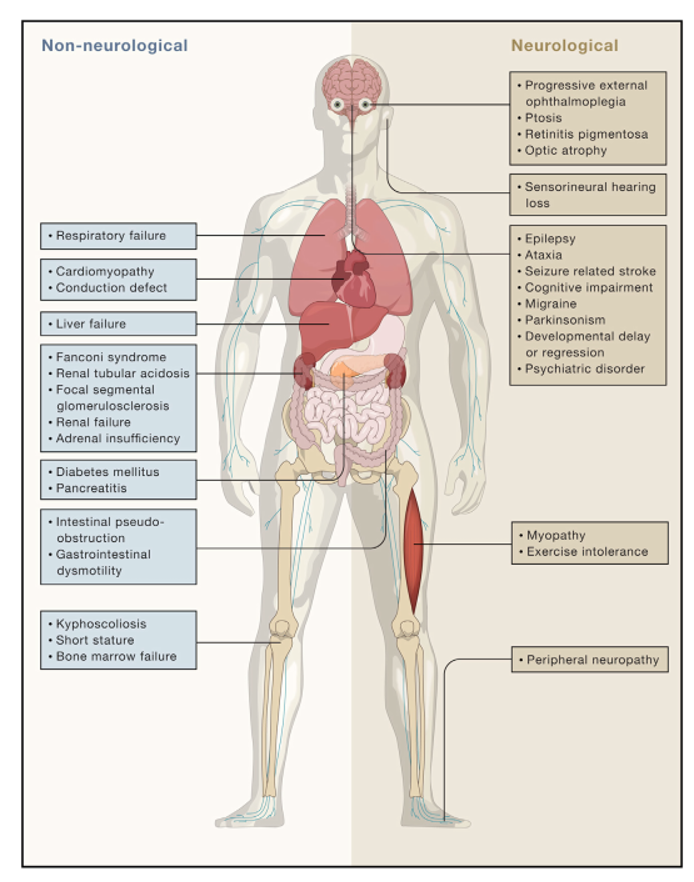
Most Common Mitochondrial Syndromes:
Leigh syndrome - Most common mitochondrial disease presentation (>80 genes). Neurological condition due to defective mitochondrial production
LHON - Leber’s Hereditary Optic Neuroretinopathy
KSS - Kearns-Sayre Syndrome
MELAS - Mitochondrial Encephalomyopathy Lactic Acidosis Stroke like episodes
MERFF - Myoclonus Epilepsy Red Ragged Fibres
NARP - Neurogenic muscle weakness Ataxia Retinitis Pigmentosa
MINGIE - Mitochondrial myopathy NeuropathyGastroIntestinaldiseaseEncephalopathy
Diagnosis:
Clinical signs
Blood and tissue histochemical and analyte measurements
Neuroimaging
Enzymatic assays of OXPHOS in tissue samples and cultured cells
DNA analysis
Low invasive biochemical investigations:
Blood/CSF lactic acid >2.1 mM
Lactic acid/pyruvate ratio
Amino Acids (e.g. alanine)
Organic acids
Muscle Histology:

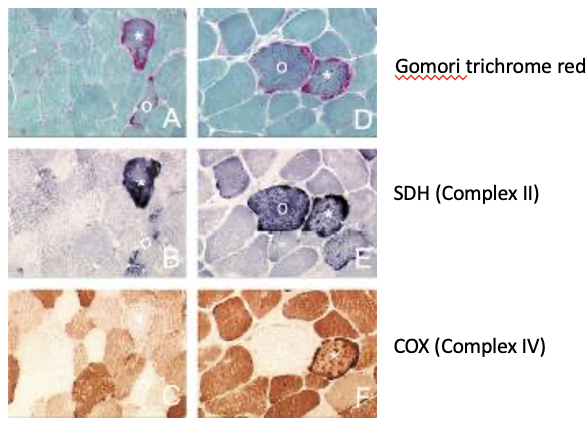
Diagnosis Summary:
Clinical clues
Characteristic syndrome/unrelated symptoms
Family history
Multiple organ involvement
Progressive
Raised lactate
COX-negative fibres on muscle biopsy
Brain MRI changes
Genetic testing (NGS)
Causes:
1. Mutations in mtDNA:
• mtDNA is maternally inherited. All mitochondria in offspring come from the mother’s egg.
• Key features of mtDNA mutations:
Heteroplasmy: Cells contain both normal and mutated mtDNA. The ratio affects disease severity.
Threshold Effect: Symptoms manifest when mutated mtDNA exceeds ~80% of total mtDNA.
- Genetic Testing:
• Next-Generation Sequencing (NGS):
• Detects mtDNA and nDNA mutations with high accuracy.
• Quantifies mutation load (heteroplasmy).
• Identifies mutations across the entire genome.
Mitochondrial Replacement Therapy
Mitochondrial Replacement Therapy (MRT) is a groundbreaking reproductive technique designed to prevent the transmission of mitochondrial DNA (mtDNA) diseases. It is also referred to as three-parent baby technology, as it involves the genetic material of three individuals. Let’s dive into its mechanisms, ethical implications, and current status.
- Purpose of MRT
• Goal: To replace defective mitochondria carrying mtDNA mutations with healthy mitochondria from a donor, thereby preventing the transmission of mitochondrial diseases.
• Target Group: Women who carry pathogenic mtDNA mutations but wish to have genetically related children free from mitochondrial disorders.
Why Is MRT Necessary?
1. Mitochondrial Diseases:
• Caused by mutations in mtDNA, which is maternally inherited.
• There are no cures for these diseases, and they can cause severe and often fatal conditions affecting the brain, muscles, heart, and metabolic systems.
Challenges of Inheritance:
• A mother with even mild heteroplasmy (a mix of normal and mutated mtDNA) can pass on severe mtDNA mutations due to random segregation during oocyte formation.
• MRT reduces this risk by replacing defective mitochondria.
Techniques in MRT
There are two primary techniques used for mitochondrial replacement:
A. Spindle Transfer
• When It’s Performed: Before fertilization.
• Process:
1. The mother’s egg is extracted, and the nuclear DNA (nDNA) is removed, leaving the defective mitochondria behind.
2. A donor’s egg (with healthy mitochondria) has its nuclear DNA removed.
3. The mother’s nDNA is inserted into the donor’s egg, which now contains the mother’s nDNA and the donor’s healthy mitochondria.
4. The reconstructed egg is fertilized with the father’s sperm and implanted into the mother.
• Outcome: The child inherits nuclear DNA from the parents and mtDNA from the donor.
B. Pronuclear Transfer
• When It’s Performed: After fertilization.
• Process:
1. Both the mother’s and donor’s eggs are fertilized with the father’s sperm, creating two zygotes.
2. The pronuclei (nuclear material from sperm and egg) from the mother’s zygote are transferred into the donor’s zygote, which has its pronuclei removed.
3. The resulting zygote contains nuclear DNA from the parents and healthy mitochondria from the donor.
4. This zygote is implanted into the mother.
• Outcome: Similar to spindle transfer, the child inherits nuclear DNA from the parents and mtDNA from the donor.
4. Advantages of MRT
1. Prevents Inheritance of mtDNA Diseases:
• Eliminates defective mtDNA, drastically reducing the risk of mitochondrial diseases.
2. Maintains Genetic Link:
• Children retain the nuclear genetic material of both parents.
3. One-Time Solution:
• Future generations are protected from mtDNA mutations since mtDNA is maternally inherited.
5. Challenges and Risks
1. Biological Risks:
• Mismatch Between Nuclear and mtDNA:
• Nuclear DNA and mtDNA have co-evolved. Introducing donor mtDNA may lead to subtle incompatibilities, potentially impacting cellular function.
• Carryover of Mutated mtDNA:
• A small amount of defective mtDNA from the mother may still be transferred, creating a risk of disease.
• Unknown Long-Term Effects:
• Since MRT is a relatively new technique, its long-term biological effects are not yet fully understood.
2. Ethical Concerns:
• Three-Parent Babies:
• Critics argue that this technology challenges traditional notions of parenthood.
• Genetic Modification:
• Although MRT does not alter nuclear DNA, some view it as a step toward germline editing.
• Access and Equity:
• MRT is expensive and may not be accessible to all families, raising concerns about healthcare inequality.
3. Regulatory Challenges:
• Different countries have varying legal and ethical stances on MRT. For example:
• The UK has legalized MRT under strict regulations.
• The US prohibits germline modification in human embryos for clinical use.
6. Current Status of MRT
1. Countries Allowing MRT:
• United Kingdom: The first country to legalize MRT in 2015. Clinics must meet stringent requirements to perform the procedure.
• Other countries considering or piloting MRT include Australia, Mexico, and some regions in Europe.
2. Successful Cases:
• In 2016, the first successful MRT baby was born in Mexico using the spindle transfer technique.
3. Research:
• Ongoing studies aim to refine MRT techniques, improve safety, and monitor long-term outcomes in children born using MRT.
7. Ethical and Societal Implications
1. Family Dynamics:
• The donor provides only mtDNA (~37 genes), which constitutes less than 1% of the total genetic material. However, the donor still contributes to the child’s biology.
2. Designer Babies?:
• Although MRT focuses on preventing disease, some fear it could pave the way for genetic enhancements in the future.
3. Informed Consent:
• Parents and donors must understand the risks, benefits, and ethical considerations before proceeding with MRT.
8. The Future of MRT
1. Improved Techniques:
• Reducing the risk of mtDNA carryover through refined methods.
2. Gene Editing Alternatives:
• Techniques like CRISPR-Cas9 might someday allow direct repair of mutated mtDNA, potentially obviating the need for MRT.
3. Broader Applications:
• MRT may eventually be explored for other reproductive challenges or mitochondrial dysfunctions.
Summary:
Mitochondrial Replacement Therapy is a transformative approach to preventing the transmission of mtDNA diseases. By replacing defective mitochondria with healthy ones from a donor, MRT offers hope to families affected by these devastating disorders. However, it raises important biological, ethical, and regulatory questions that must be carefully navigated.