Lecture 16 and 17: Chemical Reactions and Spontaneity Study Notes
Chemical Potential
Definition of Chemical Potential (μμ)
Chemical potential is a fundamental thermodynamic property that quantifies the change in a system's Gibbs free energy when an infinitesimal amount of a substance is added to the system, while keeping entropy, volume, and the amounts of other substances constant. It is essentially the partial molar Gibbs free energy of a substance.
Mathematically, for component ii in a mixture:
\mui = \left(\frac{\partial G}{\partial ni}\right){T,P,n{j \neq i}}Where G is the Gibbs free energy, ni is the number of moles of component i, T is temperature, P is pressure, and nj≠i represents the moles of all other components remaining constant.
It serves as a driving force for matter to move from regions of higher chemical potential to regions of lower chemical potential, analogous to how temperature drives heat flow or pressure drives volume changes.
For a pure substance, the chemical potential is simply its molar Gibbs free energy.
Relation of chemical potential to equilibrium constant
At equilibrium, the chemical potential of each reactant and product is constant throughout the system, and there is no net change in the amounts of species. More specifically, for a generic reaction: aA+bB⇌cC+dD
The change in Gibbs free energy for the reaction (ΔGrxnΔGrxn) can be expressed in terms of the chemical potentials of the products and reactants: \Delta G{rxn} = (c\muC + d\muD) - (a\muA + b\muB)
For a substance ii in a non-ideal mixture, its chemical potential is related to its standard state chemical potential (\mui^{\circ}) and its activity (aiai) by:
\mui = \mui^{\circ} + RT \ln a_iSubstituting this into the ΔGrxn expression at equilibrium (ΔGrxn=0):
0 = (c(\muC^{\circ} + RT \ln aC) + d(\muD^{\circ} + RT \ln aD)) - (a(\muA^{\circ} + RT \ln aA) + b(\muB^{\circ} + RT \ln aB))
0 = (c\muC^{\circ} + d\muD^{\circ} - a\muA^{\circ} - b\muB^{\circ}) + RT \ln \left(\frac{aC^c aD^d}{aA^a aB^b}\right)
0=ΔGrxn∘+RTlnKeqThus, at equilibrium, the sum of product chemical potentials (weighted by stoichiometric coefficients) equals the sum of reactant chemical potentials.
Equilibrium binding of molecular oxygen to hemoglobin
Hemoglobin (Hb) is a protein in red blood cells responsible for transporting oxygen. Its ability to bind oxygen is a classic example of chemical equilibrium in biological systems, which is governed by chemical potentials.
The binding can be represented as:
Hb(aq)+O2(g)⇌HbO2(aq)The driving force for oxygen to bind to hemoglobin is the difference in chemical potential of oxygen in the gas phase (lungs) and in the bound state with hemoglobin. High oxygen partial pressure in the lungs leads to higher oxygen chemical potential, favoring binding to Hb. In tissues, where oxygen partial pressure is lower, the chemical potential of bound oxygen becomes relatively higher, leading to its release.
This binding is cooperative, meaning the binding of one O2 molecule to hemoglobin increases the affinity for subsequent O2 molecules, which can be understood more deeply through changes in conformational states and their respective chemical potentials.
Reaction Spontaneity
General Equation for Reaction Spontaneity
For any reaction system not necessarily at equilibrium, the change in Gibbs free energy for a reaction (ΔGrxn) determines its spontaneity under constant temperature and pressure conditions. A reaction is spontaneous if it proceeds towards lower Gibbs free energy.
The general equation for reaction spontaneity is:
ΔGrxn=ΔG∘rxn+RTlnQTheoretical Explanation: This equation relates the Gibbs free energy change of a reaction at any given set of conditions (ΔGrxn) to its standard Gibbs free energy change (ΔG∘rxn) and the reaction quotient (Q).
The ΔGrxn∘ term accounts for the intrinsic thermodynamic favorability of the reaction under standard conditions (1 bar partial pressure for gases, 1 M concentration for solutes, pure liquids/solids, all at a specified temperature, usually 298.15 K).
The RTlnQ term accounts for the effect of non-standard concentrations or pressures of reactants and products. The system will naturally tend to adjust concentrations to reduce ΔGrxn. R is the ideal gas constant (8.314J⋅mol−1⋅K−18.314J⋅mol−1⋅K−1), and T is the absolute temperature in Kelvin.
Detailed Breakdown of Terms:
ΔGrxn: Gibbs free energy change for the reaction under the current, non-standard conditions. This value determines the actual spontaneity and direction of the reaction.
ΔG∘rxn: Standard Gibbs free energy change, specific for the reaction under standard conditions. It is calculated from the standard Gibbs free energies of formation (ΔG∘f) of products and reactants:
ΔG∘rxn=∑iniΔG∘f,products,i−∑jmjΔGf,reactants,j∘Where ni and mj are the stoichiometric coefficients for products and reactants, respectively.
Example Calculation for Standard Free Energy:
Consider the reaction: 2SO2(g)+O2(g)⇌2SO3(g)To calculate ΔG∘rxn, you would typically use tabulated values:
ΔG∘f(SO2(g))=−300.1kJ/molΔG∘f(O2(g))=0kJ/mol (element in standard state)
ΔG∘f(SO3(g))=−371.1kJ/mol
ΔG∘rxn=[2⋅ΔG∘f(SO3)]-[2⋅ΔG∘f(SO2)+1⋅ΔG∘f(O2)]
ΔG∘rxn=[2(−371.1)]−[2(−300.1)+1(0)]
ΔGrxn∘=−742.2−(−600.2)=−142.0kJ/mol
The Reaction Quotient (Q): This term quantifies the relative amounts of products and reactants at any given point in time (not necessarily equilibrium). It has the same mathematical form as the equilibrium constant but uses non-equilibrium activities.
Q=∏iainiWhere ai is the activity of species i and ni is its stoichiometric coefficient. For gases, activity is often approximated by partial pressure in bar; for solutes, by molar concentration in mol/L; for pure solids/liquids, activity is defined as 1.
Uses an ICE table for calculations: An ICE (Initial, Change, Equilibrium) table is a systematic way to track concentrations (or partial pressures or activities) of reactants and products during a reaction. However, for calculating Q, you only need the initial or current concentrations/pressures to plug into the Q expression.
Example using ICE Table for Q (pre-equilibrium):
Consider again: N2O4(g)⇌2NO2(g)Suppose initially we have 1.0 atm N2O4 and 0.5 atm NO2.

The current activities (approximated by partial pressures in bar, assuming 1 atm \approx 1 bar):
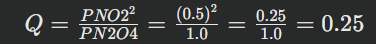
This value of Q would then be used in the general spontaneity equation.Key concepts of spontaneity
The sign of ΔGrxn directly indicates the spontaneity of a reaction at constant T and P:
ΔGrxn=0: the reaction mixture is at equilibrium.
Explanation: When ΔGrxn=0, the system has reached a state of minimum Gibbs free energy. There is no net driving force for the reaction to proceed in either the forward or reverse direction. The rates of the forward and reverse reactions are equal. At this point, the reaction quotient Q becomes equal to the equilibrium constant Keq, leading to:
0=ΔG∘rxn+RTlnKeq ⟹ ΔG∘rxn=−RTlnKeq
ΔGrxn<0: forward reaction is spontaneous.
Explanation: A negative ΔGrxn indicates that a net decrease in Gibbs free energy will occur if the reaction proceeds in the forward direction. This means the products are favored over the reactants under the current conditions, and the reaction will spontaneously move towards forming more products until equilibrium is reached.
ΔGrxn>0: reverse reaction is spontaneous.
Explanation: A positive ΔGrxn means that the system's Gibbs free energy would increase if the reaction were to proceed in the forward direction. Since systems naturally move towards lower energy, the forward reaction is non-spontaneous. Instead, the reverse reaction (reactants forming from products) would be spontaneous, as it would lead to a decrease in Gibbs free energy until equilibrium is established.
Calculating Reaction Direction
Calculate ΔGrxn∘ using tables of standard thermodynamic quantities:
ΔGf∘ (Standard Gibbs Free Energy of Formation): This is the Gibbs free energy change when 1 mole of a compound is formed from its constituent elements in their standard states. Tabulated values for many compounds are available.
ΔH∘f (Standard Enthalpy of Formation) and S∘ (Standard Molar Entropy): If ΔG∘f values are not directly available, you can calculate ΔH∘rxn and ΔS∘rxn from standard enthalpies of formation and standard molar entropies, respectively. Then, use the fundamental relationship: ΔG∘rxn=ΔH∘rxn−TΔSrxn∘
Step-by-step calculation example: For 2SO2(g)+O2(g)⇌2SO3(g), in addition to the previous ΔG∘fΔG∘f values, suppose we have:
ΔH∘f(SO2(g))=−296.8kJ/molS∘(SO2(g))=248.2J/(mol⋅K)
ΔH∘f(O2(g))=0kJ/mol
S∘(O2(g))=205.1J/(mol⋅K)
ΔH∘f(SO3(g))=−395.7kJ/mol
S∘(SO3(g))=256.8J/(mol⋅K)
Step 1.1: Calculate ΔH∘rxnΔH∘rxn:
ΔH∘rxn=[2(−395.7)]−[2(−296.8)+1(0)]=−791.4−(−593.6)=−197.8kJ/molStep 1.2: Calculate ΔS∘rxn: (Careful with units, convert J to kJ)
ΔS∘rxn=[2(256.8)]−[2(248.2)+1(205.1)]ΔS∘rxn=513.6−(496.4+205.1)
ΔS∘rxn=513.6−701.5
ΔS∘rxn=−187.9J/(mol⋅K) or −0.1879kJ/(mol⋅K)
Step 1.3: Calculate ΔG∘rxnΔG∘rxn at 298.15 K:
ΔG∘rxn=ΔH∘rxn−TΔS∘rxn
ΔG∘rxn=−197.8kJ/mol−(298.15K)(−0.1879kJ/(mol⋅K))ΔG∘rxn=−197.8kJ/mol+56.04kJ/mol=−141.76kJ/mol
(This value is close to the one calculated directly from ΔG∘f values, indicating consistency. Differences may arise from rounding or different data sources.)
Determine the starting activities using initial conditions provided in the problem
This involves setting up the Reaction Quotient (Q) expression using the given initial concentrations or partial pressures. Remember that activities are derived from these, and generally:
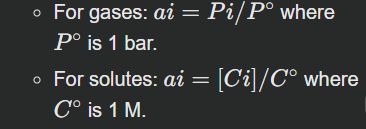
For pure solids/liquids: ai=1 (their concentration doesn't change significantly during the reaction).
Step-by-step example: For N2O4(g)⇌2NO2(g) at 298 K, assume initial partial pressures are PN2O4=0.5 bar and PNO2=0.1 bar.
(Given ΔG∘rxn=5.4kJ/mol at 298 K from context).
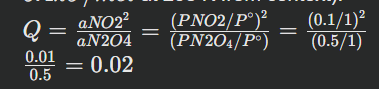
Use the equation ΔGrxn=ΔG∘rxn+RTlnQ to identify the direction of change and determine activities in equilibrium mixture
Substitute the calculated ΔG∘rxn and Q into the equation to find ΔGrxn, then interpret its sign. The RTlnQ term quantifies the adjustment from standard conditions to current conditions.
Continuing the example:
R=8.314J/(mol⋅K)=0.008314kJ/(mol⋅K)R=8.314J/(mol⋅K)=0.008314kJ/(mol⋅K)
\Delta G{rxn} = 5.4 \text{ kJ/mol} + (0.008314 \text{ kJ/(mol\cdot K)})(298 \text{ K}) \ln(0.02) ΔGrxn=5.4+(2.479)ln(0.02)ΔGrxn=5.4+(2.479)ln(0.02)
ln(0.02)≈−3.912ln(0.02)≈−3.912
ΔGrxn=5.4+(2.479)(−3.912)ΔGrxn=5.4−9.70=−4.30kJ/mol
Interpretation: Since ΔGrxn=−4.30kJ/mol (which is negative), the reaction is spontaneous in the forward direction under these initial conditions. This means it will proceed to form more NO2 and consume N2O4 until equilibrium is reached.
Reaction Quotient (QQ): Same form as equilibrium constant but activities not necessarily for an equilibrium mixture
As introduced, Q is a measure of the relative amounts of products and reactants present in a reaction at any given time. It is calculated using the same formula as the equilibrium constant (Keq), but with the current activities (ai) rather than equilibrium activities.
Example: For aA+bB⇌cC+dD, the reaction quotient is:
This unitless value allows us to compare the current state of the reaction mixture to its equilibrium state by comparing Q to Keq.
Standard Free Energy Change in Chemical Reactions
Interpretation of ΔGrxn∘
The standard free energy change (ΔGrxn∘) describes the Gibbs free energy change for a reaction when all reactants and products are in their standard states.
Standard State Conditions Defined:
For a gas: Partial pressure of 1 bar.
For a solute in solution: Concentration of 1 M (mol/L).
For a pure liquid or solid: The pure substance itself at 1 bar pressure.
For an element: Its most stable allotropic form at 1 bar and specified temperature (usually 298.15 K).
Standard free energy change can be interpreted in two scenarios:
When assuming activity = 1 for all species:
The interpretation of ΔG∘rxn is effectively the ΔGrxn for a hypothetical process where 1 mole of reactants in their standard states are completely converted to 1 mole of products in their standard states. This holds true because if all activities (ai) are 1, then the reaction quotient Q becomes 1. Q≡1→ΔGrxn=ΔG∘rxn+RTln(1)=ΔG∘rxn+0=ΔGrxn∘
Reminder: Activity of a pure component is 1: This means that when a substance is in its pure form (e.g., solid ice at 0 °C, pure liquid water), its activity is defined as 1 in its standard state. This simplifies calculations as these terms drop out of the Q (and Keq) expression