M1L8 Epigenetic regulation and DNA integrity
Epigenetics - the structural adaptation of chromosomal regions so as to register, signal or perpetuate altered activity states
Transcription factors are key for gene expression and can be used to reprogram cells (eg. using Yamanaka factors OCT4, DOX2, c-MYC, KLF4 to reprogram fibroblasts into stem cells)
This process is highly inefficient due to organisation of DNA into chromatin (heterochromatin and euchromatin)
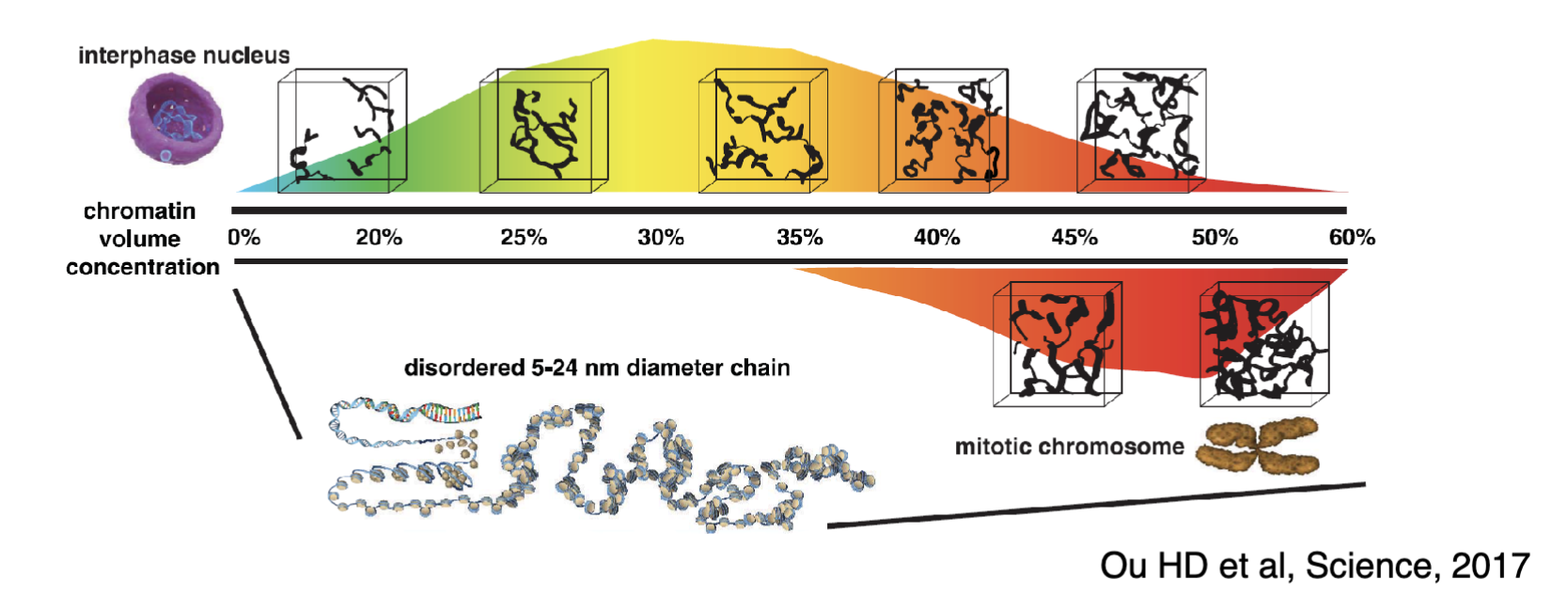
Chromatin is organised into nucleosomes (octamer consisting of two each of H2A, H2B, H3 and H4 with DNA wrapped around)
Nucleosomes (beads on a string) are further folded many times into solenoid structure, fibres, chromonema, chromatids and finally mitotic chromosomes
However new evidence shows there is no discrete orders of chromatin structures and chromatin is folded into a variety of compacted states in no particular order
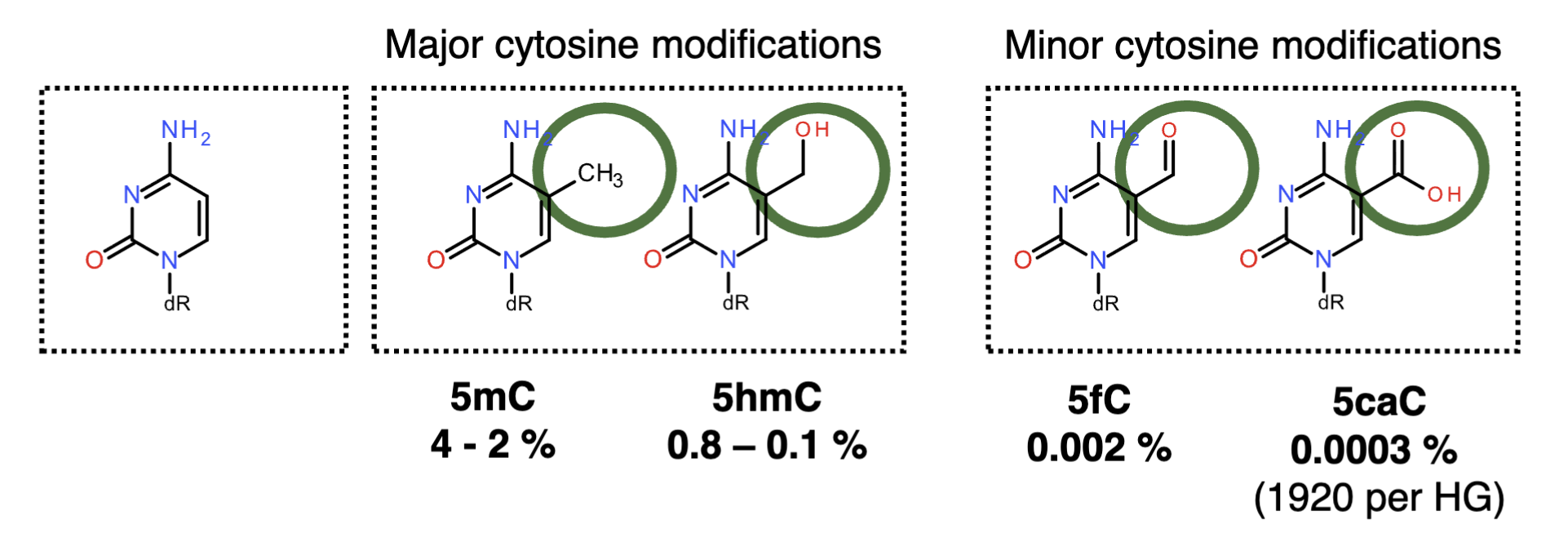
Cytosine tends to get modified, forming 5mC (4-2% of genome) which can be further modified to 5hmC (0.8-0.1%)
Minor modifications - 5fC (0.002%), 5caC (0.0003%)
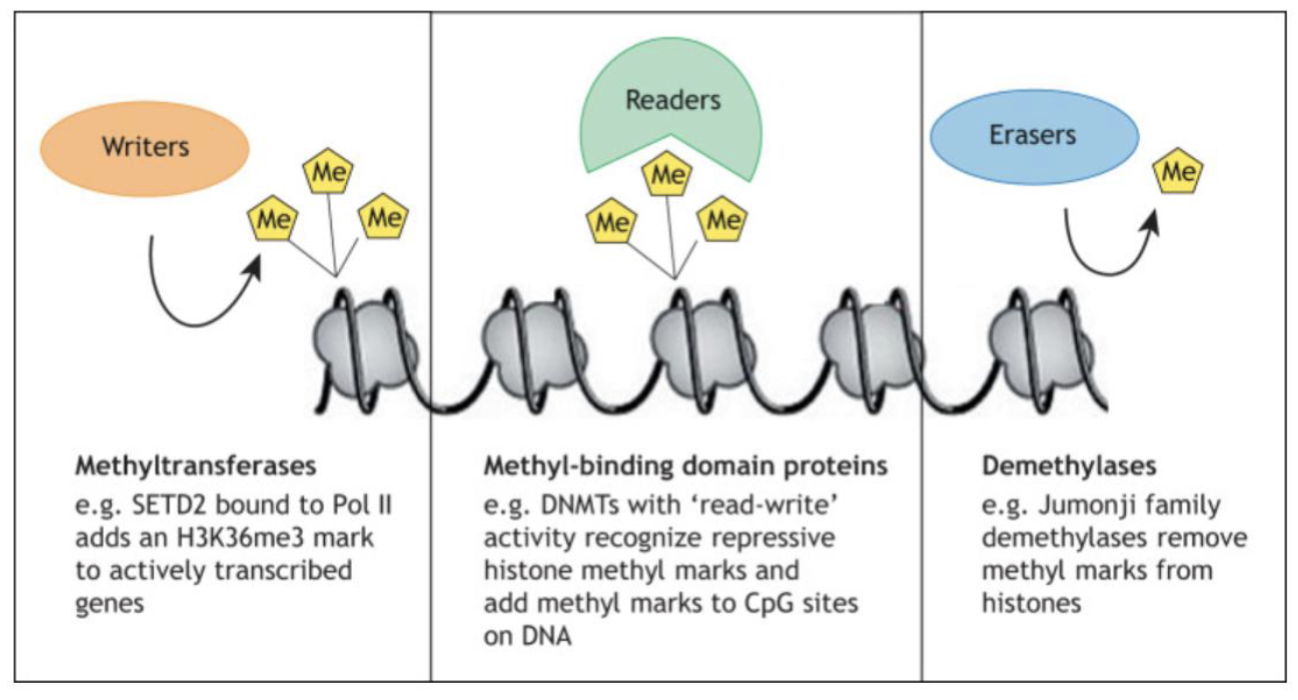
N terminal tails of nucleosomes can be chemically modified - methylation, acetylation, phosphorylation, ubiquitylation…
Writers (eg. methyltransferase), readers (eg. methyl-binding domain proteins), erasers (eg. demethylases)
Histone variants may also be produced and substitute canonical forms of histones in the nucleosome
C terminus tail of H2AX is longer than canonical H2A which can receive modifications
N terminus is highly conserved
γ-H2AX (phosphorylation of p-Ser139 by ATM) signals DSBs and recruits other proteins for repair
Transcription
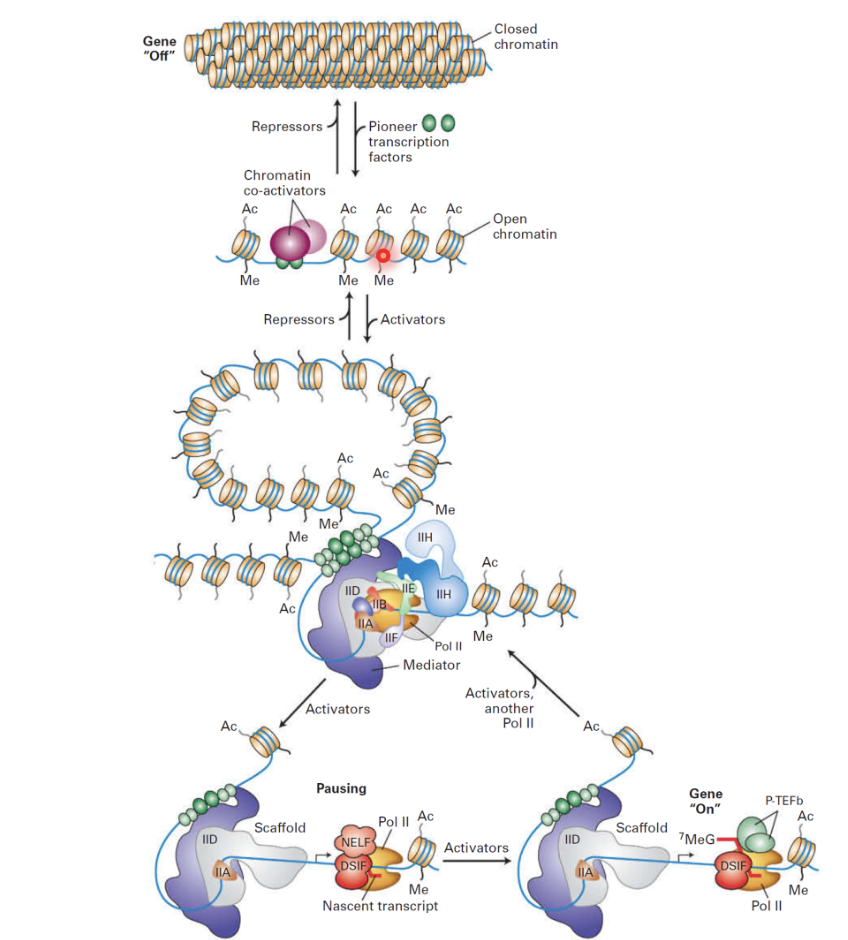
Pioneering TFs can invade inaccessible sites (repressed state is modulated by repressors, though there are few relative to activators/co-activators and it is the default): promoters and enhancers
Activators and co-activators open up chromatin structure
Methylation and acetylation marks modulate chromatin remodelling to regulate the permissibility of the gene promoter for transcription
Enhancers and promoters interact via mediator complex
Enhancers can be brought into the vicinity of the promoter to form the transcription pre-initiation complex with RNA pol II and TFs
RNA pol II can be paused and released resulting in a burst of transcription
RNA pol II can start transcribing and then it pauses
Pause mechanism is associated with NELF and DSIF (protein complexes that work together to cause promoter-proximal pausing)
Additional signals needed for RNA pol II to get released and replace NELF with P-TEFb for productive elongation
Increased concentration of activators increases frequency of bursts of transcription to increase transcription rate
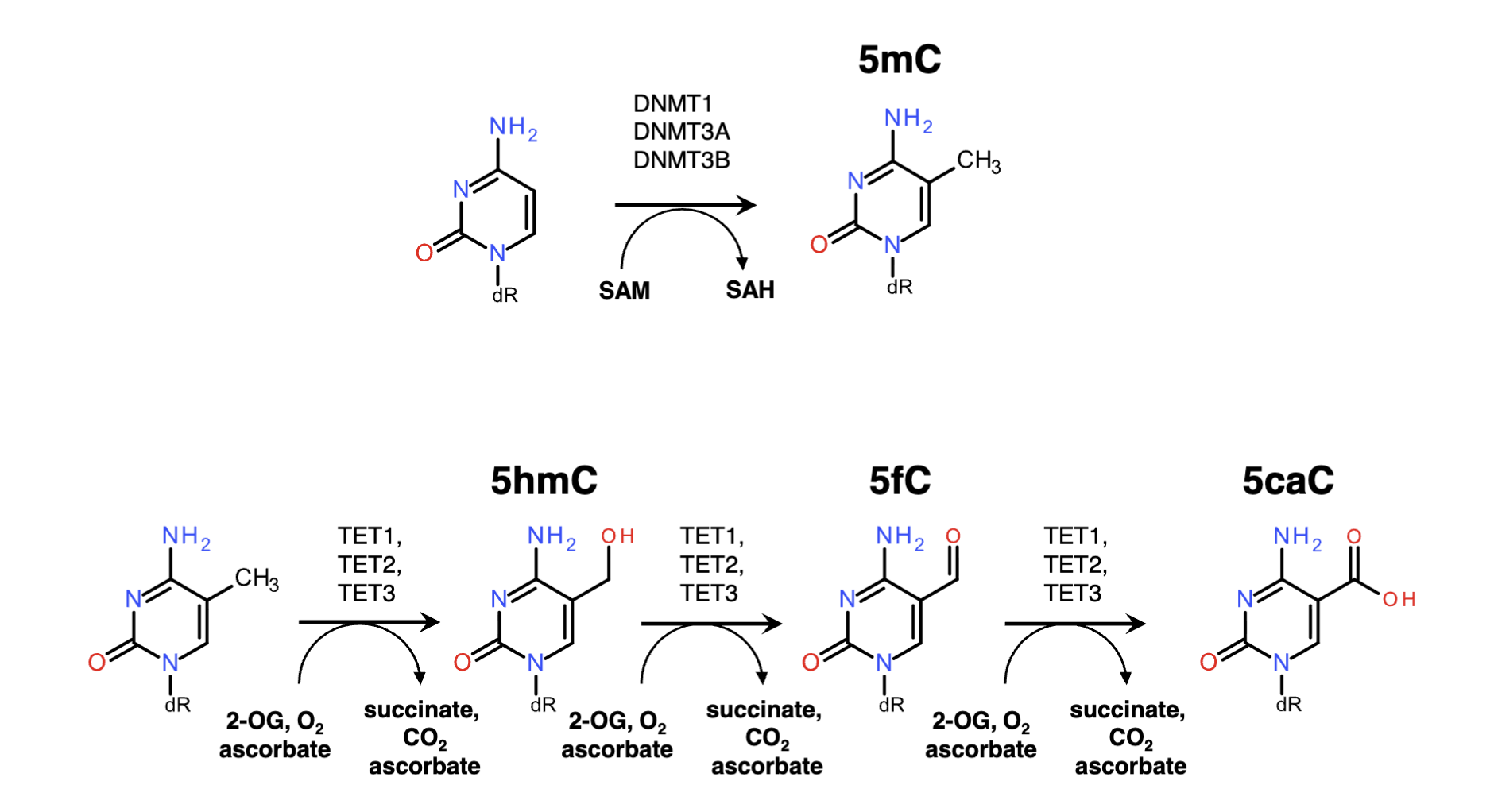
DNA modifying enzymes
Cytosine can by methylated by DNMT1, DNMT3A, DNMT3B, which converts SAM to SAH to produce 5mC
5mC is converted to 5hmC using TET1, TET2, TET3 (while 2-OG, O2 ascorbate —> succinate, CO2 ascorbate), which can further convert it to 5fC, then 5caC
Patterns of 5mC
C in CpG is methylated and it is found in a symmetrical in both strands of DNA
Bimodal distribution of methyl CpG in the genome
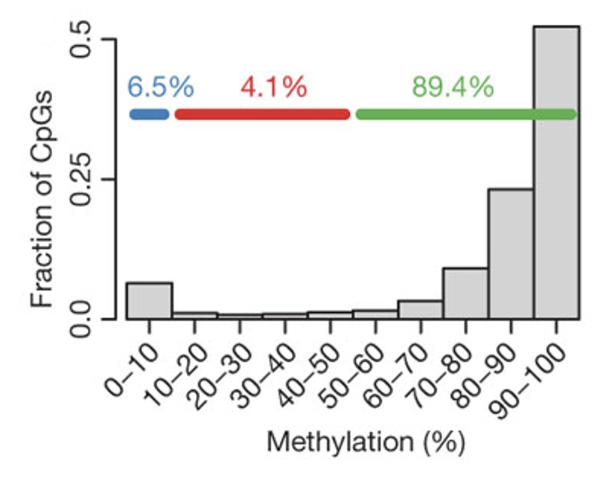
One peak at heavily methylated CpGs (found in intergenic DNA and repetitive elements which are transcriptionally silent) and another at unmethylated CpGs (found in CpG islands and active promoters which are transcriptionally active)
CpG islands have very little modification (mostly unmethylated regulatory hubs) and are located in the TSS and active enhancers, whereas the most of the rest of the genome is methylated
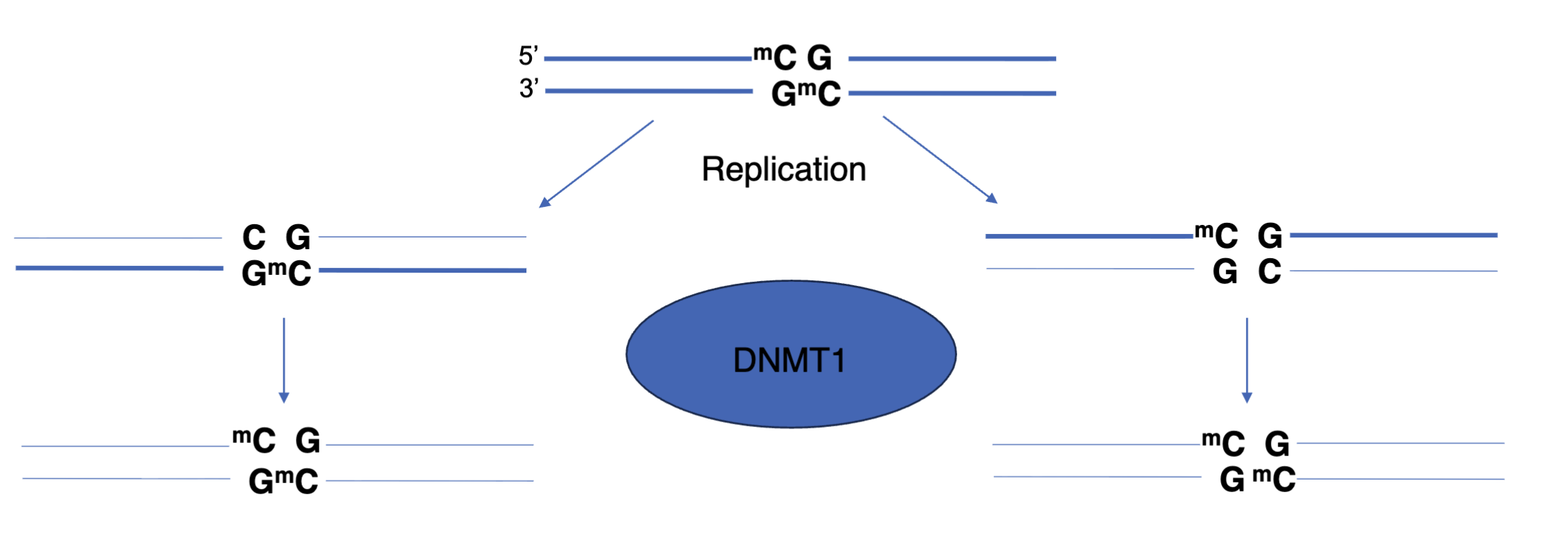
Inheritance of DNA methylation
Due to semi conservative replication the newly synthesised strand is not methylated (hemimethylation)
DNMT1 restores methylation
PCNA can interact with DNMT1 to restore methylation patterns
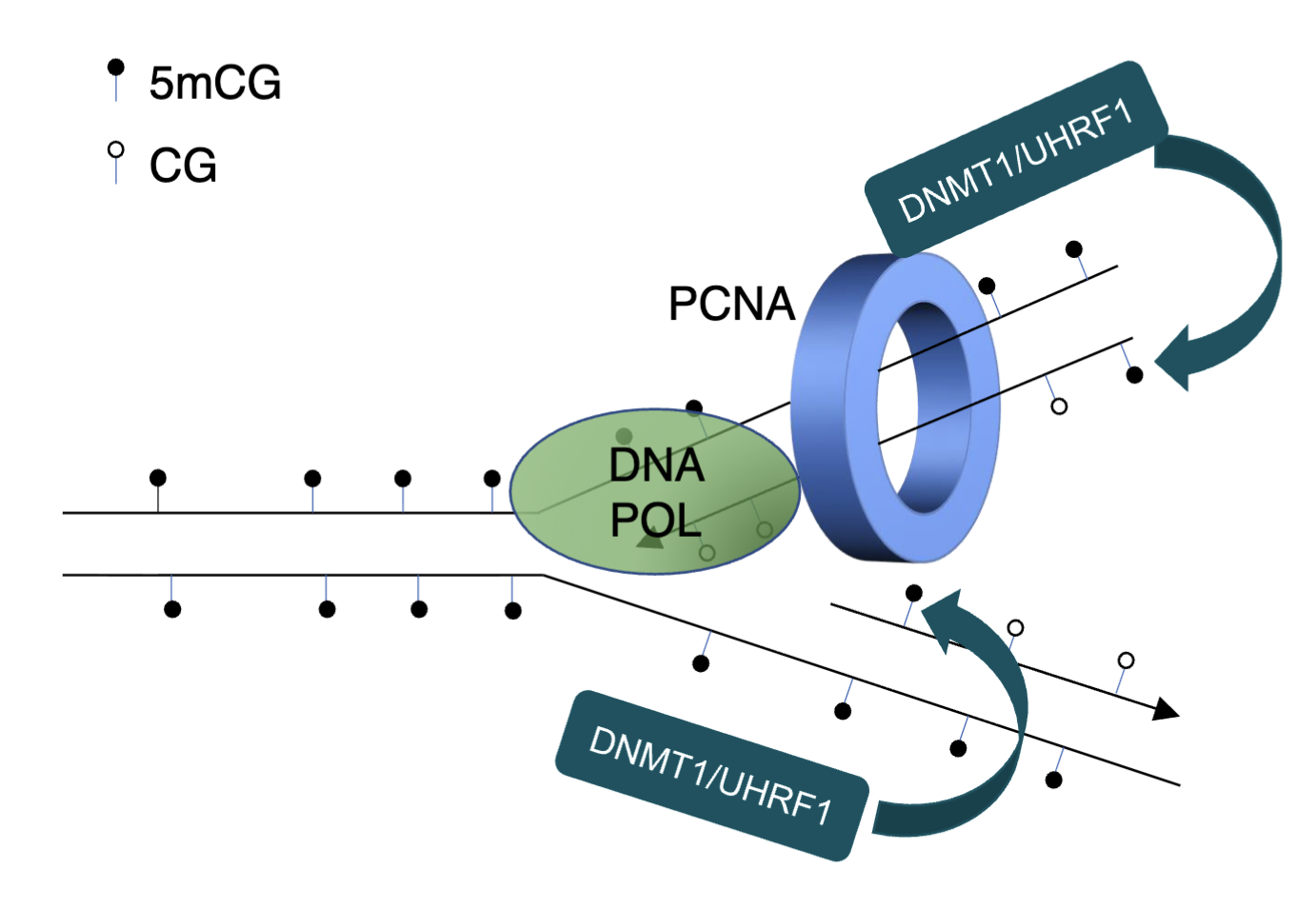
DNMT1 has a CXXC domain which binds to unmethylated CpG with high affinity which positions catalytic TRD domain away from cytosine, preventing it from depositing methylation
BAH2 loop restrains TRD in retracted position
Autoinhibitory linker occludes catalytic site
Protects unmodified CpGs from DNMT1
Demethylation by TET enzymes
5mC is oxidised to 5hmC and further to 5fC and to 5caC
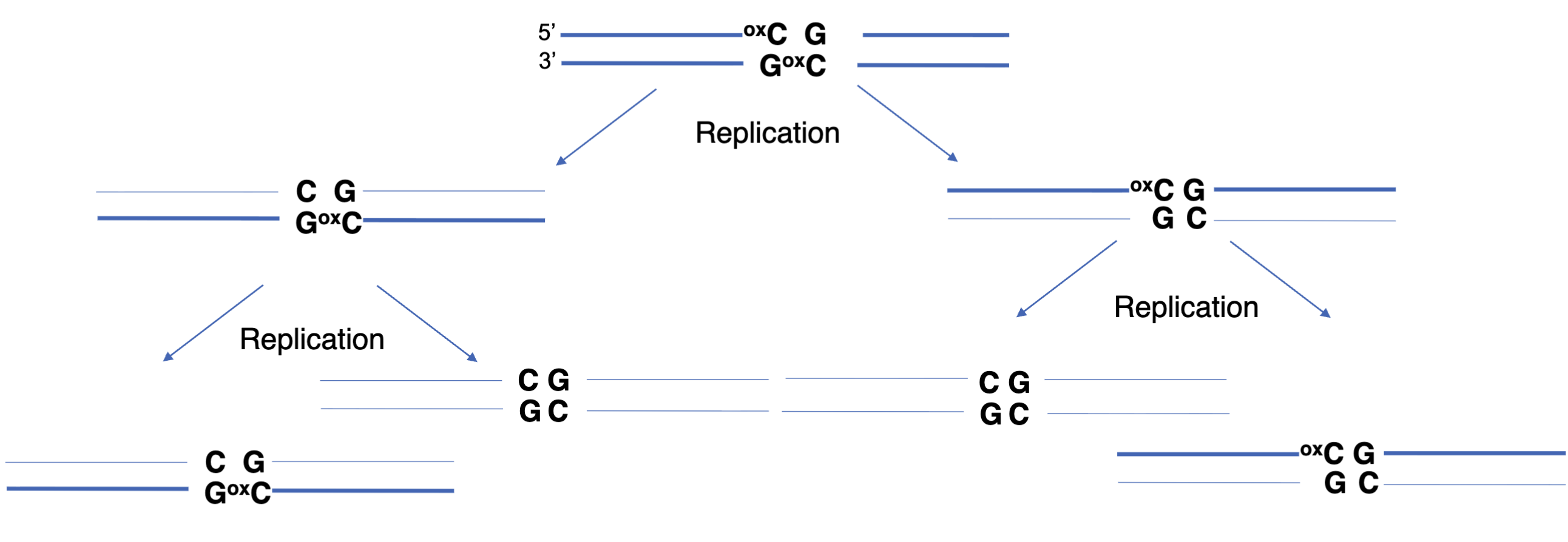
Passive (replication-dependent) loss of methylation (dominant)
Oxidised cytosine is lost due to semi conservative replication as hemioxidised CpG is a poor substrate for DNMT1
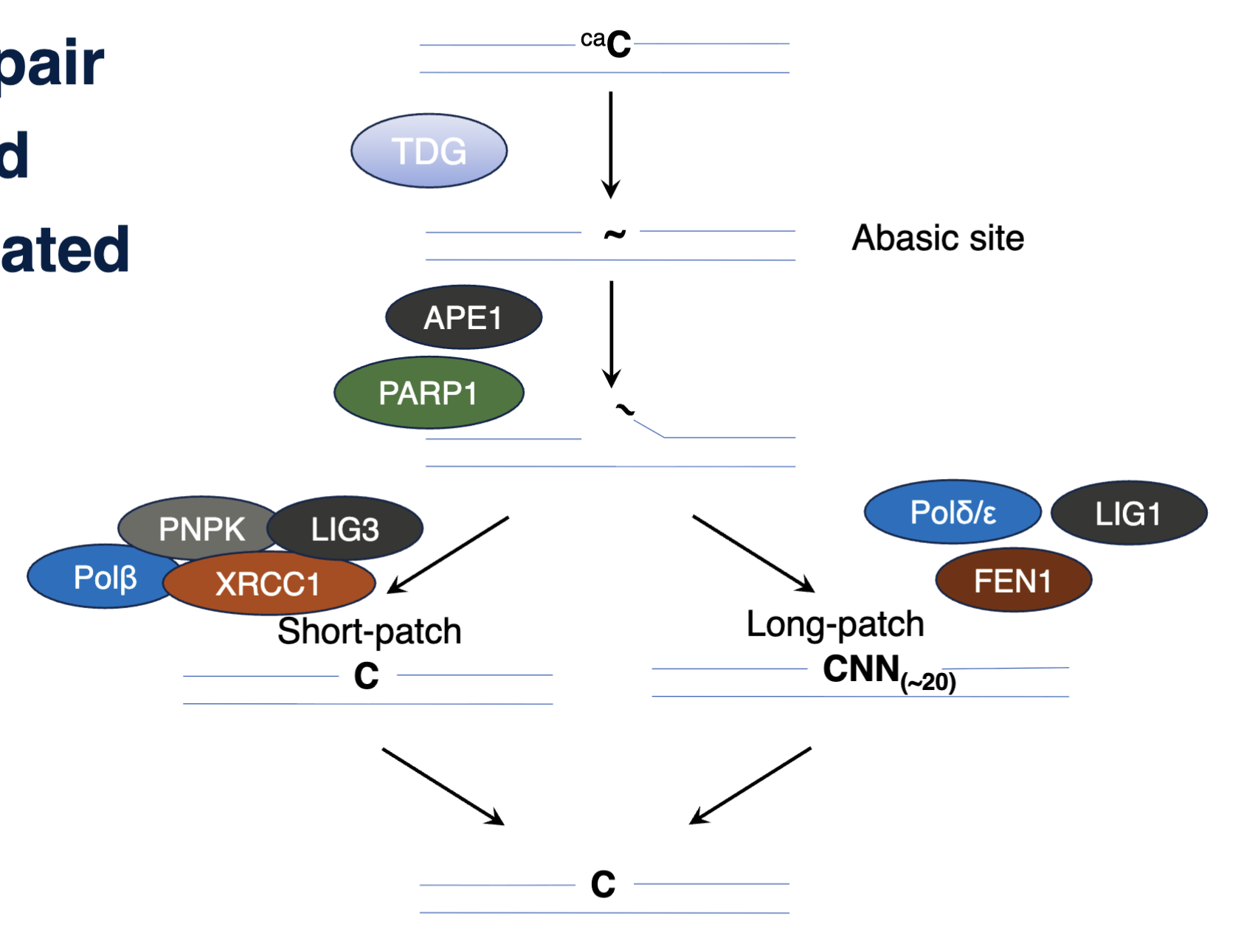
BER mediated demethylation
TDG removed caC, forming an abasic site, cleaved by APE1 and PARP1, initiating short patch or long patch repair
Readers of CpGs
5mC has high affinity to proteins with methyl CpG binding domains and zinc finger domains
Unmethylated CpGs are recognised with proteins with CXXC domain, including DNMT1, other chromatin modifiers, and TET enzymes which hydroxylate 5mC
5mC in promoters in promoters inhibit transcription and silence gene expression
CpG islands at TSS are usually maintained in unmethylated state
CpG island methylated phenotype (CIMP) in cancer
Testis expressed genes not expressed in any other cells but sometimes expressed in tumour cells due to DNA methylation remodelling
Imprinted genes - ~200 genes that remember whether they have been inherited from mother or father due to methylation mark left by parental germline during gametogenesis
Imprinted alleles are monoallelically expressed, one is methylated/silenced and the other is unmethylated/active
Genes on inactive X chromosome - in females one of the two X chromosomes is silenced
In embryonic development one X chromosome is randomly selected to be inactivated
XIST gene of that chromosome is activated and produces lncRNA that coats the chromosome in cis. recruited silencing machinery
DNA methylation of promoter CpG islands acts as an epigenetic lock to maintain silencing through cell divisions
Retrotransposons (interspersed repetitive elements) are silenced by methylation
Genetic disorders
Germline - mutation in DNMT3B in immunodeficiency-centromeric instability facial anomalies syndrome 1 (ICF1)
Somatic - mutation in DNMT3A in AML
Germline mutation in MECP2 (methylation reader) - Rett syndrome
Common alterations in cancer
Hypomethylation of centromeric sequences
Genomic instability due to hypomethylation
CpG island hypermethylation (CIMP)
Loss of 5hmC
DNA damage
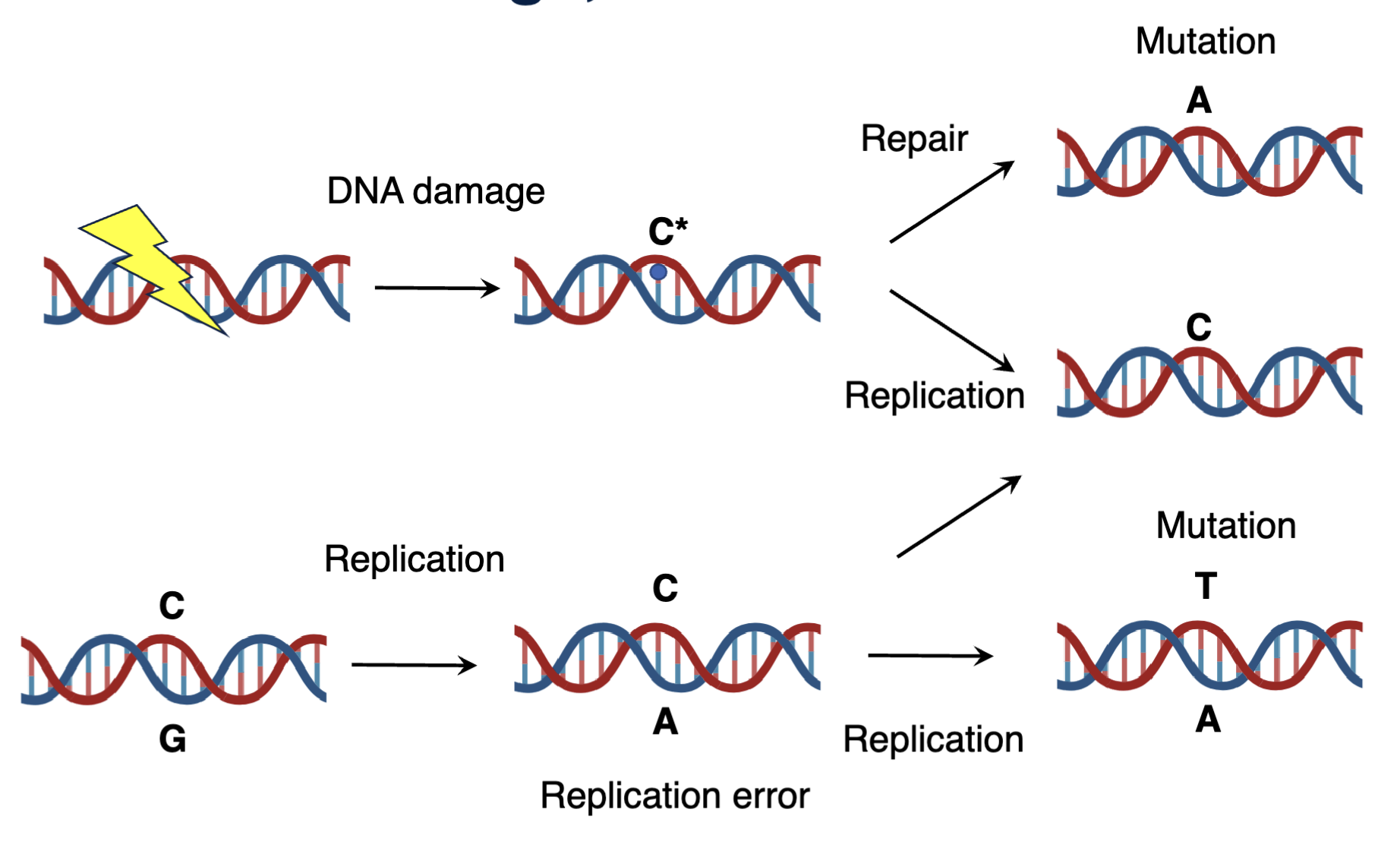
Damage product can be repaired into the native state or replicated into a mutation which are hard to later repair as it is difficult to discern the context
Mutational signatures - documented mutations are being mathematically organised into signatures
Mutational signatures of mutagens
Rogue activity of DNMTs or TETs
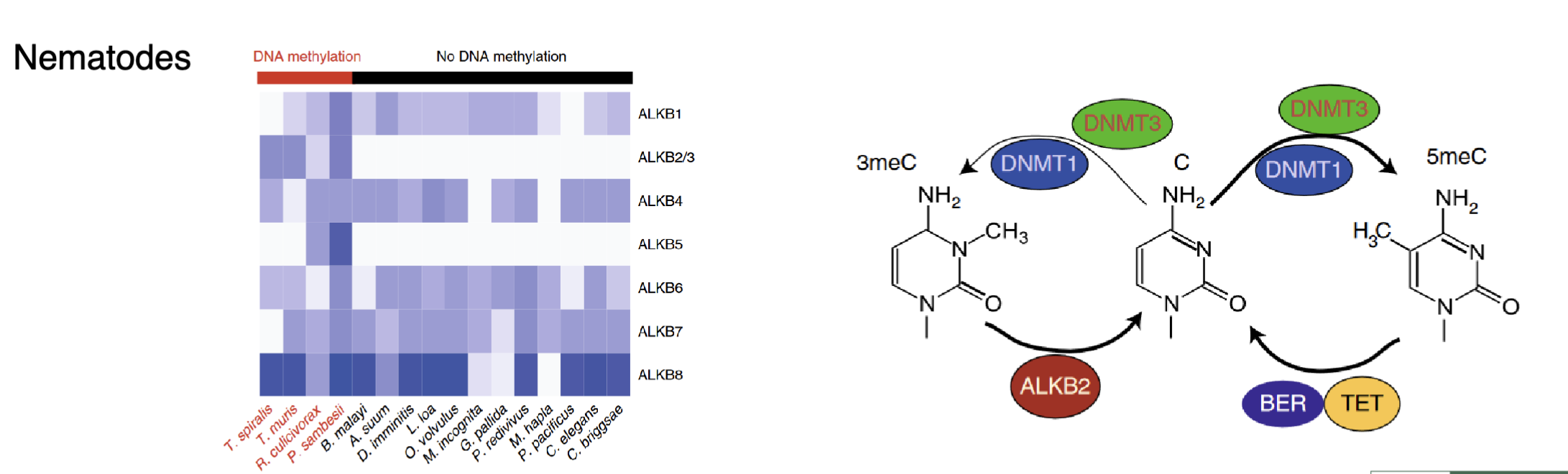
In nematodes AlkB2/3 and AlkB5 are always present if their genome contains methylation
Sometimes instead of methylating the 5’ position DNMTs methylate the wrong group (eg 3meC) which can affect base pairing and cause mutations
AlkB family of dioxygenases are repair enzymes that can remove the undesired methyl group
5mC involved in cyclobutane pyrimidine dimer (CPD) formation after UV
Can be repaired by global genome NER (GG-NER) or transcription coupled NER (TC-NER)
Investigating mutations
7 out of 8 most commonly mutated residues in TP53 are C>T at commonly methylated CpG sites
43% germline mutations in MECP2 are C>T at commonly methylated CpG sites
~70% of tumours have SBS1 mutational signature
Spontaneous deamination
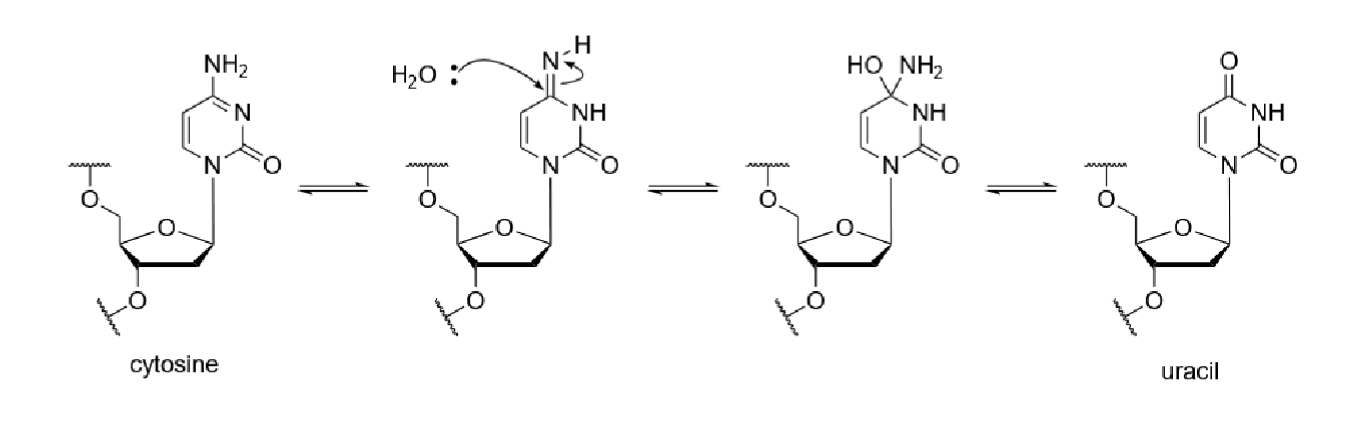
Cytosine can react with water to be deaminated and converted to uracil by losing an amino group
5mC can be converted into thymine
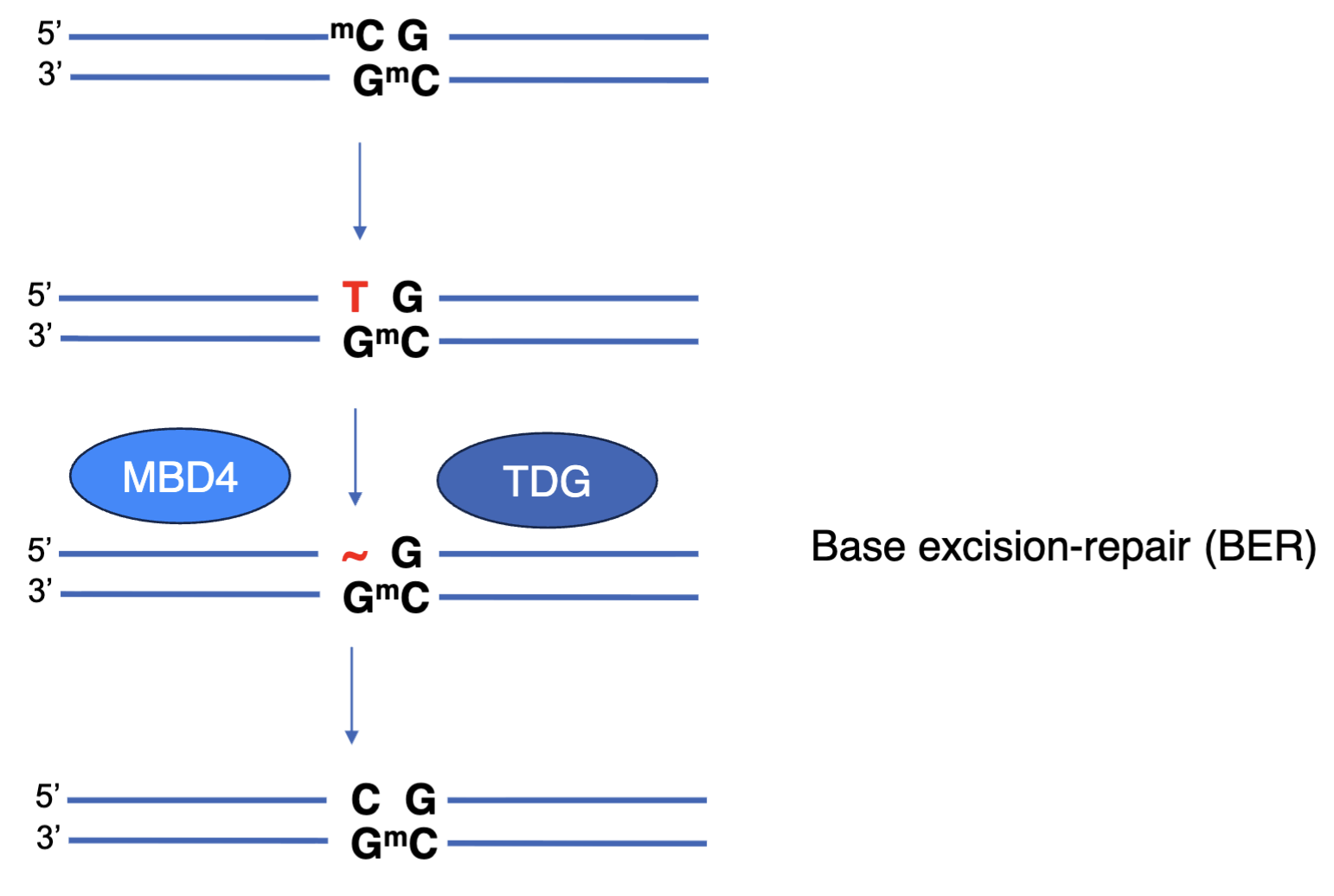
TDG and MBD4 can use BER to repair T:G mismatches caused by chemical modification
5mC can be converted to T
5mC more mutable than C or 5hmC
Brain develops more 5hmC to protect against mutation
Deamination rate of 5hmC to 5hmU is similar to unmodified C, whereas deamination rate of 5mC is notably higher
Hypermutated cancers have defective DNA pol ε or MMR enzymes
DNA pol ε can replicate DNA using its polymerase domain and also has proofreading activity due to exonuclease domain
Mutations in exonuclease domain can increase mutations when replicating the genome
This has shown to increase mutability of CpGs, predominantly in the leading strand because the lagging strand synthesis uses Pol δ which has its own proofreading domain
Hypotheses explaining these findings:
Pol ε and MMR are involved in non-canonical repair of mismatches generated by spontaneous deamination
Deamination is too slow to explain the observed mutation rate
Pol ε and MMR mutations produce gain of function mutator phenotype - the defective enzymes may introduce errors or misprocess damaged bases, becoming a source of mutations rather than just failing to fix them
Shared mutation patterns across cancers - plausible
Pol ε has low fidelity when incorporating nucleotides opposite 5mC
CpG mutation enrichment and leading strand asymmetry makes this most consistent with the data
If mismatches escape proofreading activity of DNA pol, MMR can be activated
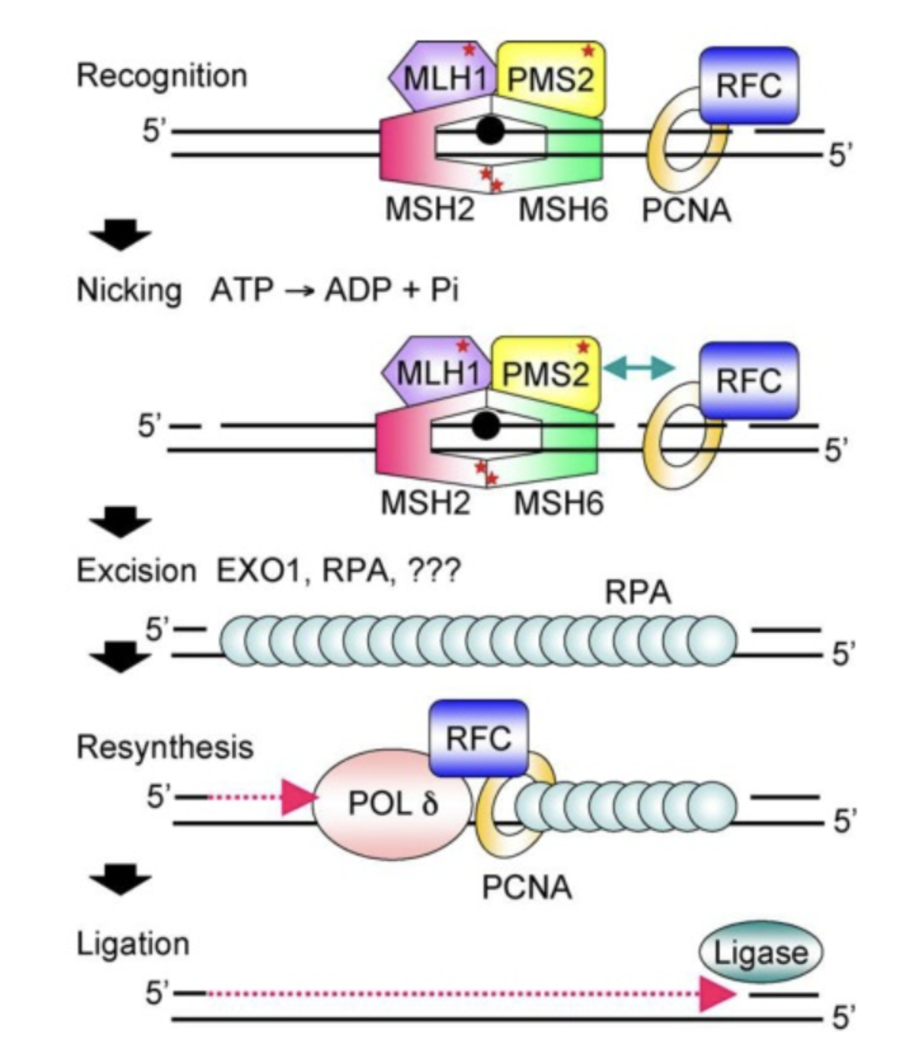
MutS homologues detect mismatch and form a tertiary complex with MutL homologues and the mismatch
PMS2 is a mismatch-activates strand-specific nuclease which creates a break in the strand containing a nick provided
The nick is either the discontinuously synthesised Okazaki fragments which acts as the signal for the newly synthesised strand on the lagging strand, or it is created by PCNA which directs PMS2 using its endonuclease activity to cut the strand being synthesised
The nick could also be from the ribonucleotide excision repair system from RNase H2 if the mismatched nucleotide is a ribonucleotide
EXO1 excises the mismatch and the gap is filled by PCNA, Pol δ and DNA ligase
Measuring mutations
Purified Pol E complex (WT or mut) incubated with methylated or nonmethylated DNA
Polymerase error sequencing (PER-seq)
Single molecule barcoding - 19N barcode for each of the original molecules
Linear amplification of original molecule - propagates any true replication errors introduced by Pol ε when incubated with original template but new mutations introduced by Pol ε during amplification are not replicated
In linear amplification each original DNA molecule acts as a template for only 1 strand per replication cycle and the new strand does not become a template for further amplification
Reads sharing the same barcode are aligned and compared to identify true mismatches introduced by Pol ε in the incubation
Allows mismatches to be quantified - separate sequencing of top and bottom strands allows subtraction of background errors produced in template
Observed that mutated Pol ε does produce higher CpG mutations and if there are methylated CpGs in the parent strand then more mutations are generated
If there is methylation on other positions there are less mutations, suggesting 5mC is particularly associated with low fidelity of Pol ε