Exam 2 IBGS 511
Soto 4 10/12 DNA Damage and Repair
Endogenous damage: Damage caused by normal cellular processes
Examples:
• Hydrolytic depurination
• Hydrolytic deamination
• Oxidation
• Alkylation
• Polymerase incorporation errors
Exogenous damage: Damage caused by external factors
Examples:
• UV from sunlight
• Ionizing radiation
• Environmental toxins (e.g. cigarette smoke, cytotoxic
chemotherapy agents)
Types of DNA damage (2)
Hydrolytic Endogenous DNA Damage: Damage from water-hydrolysis
• take place on double-helical DNA, neither break the phosphodiester backbone
Ex:
Depurination; remove a guanine of adenine from DNA
• can lead to a loss of nucleotide pair
Deamination; Cytosine converts to uracil (U:G mispair)
• [adenine → hypoxanthine] [ guanine → Xanthine] [5-Methylcytosine → Thymine]
• results in the substitution of one base for another when the DNA is replicated.
Oxidative Endogenous DNA damage: Damage from reactive oxygen species (ROS) - oxidation
• byproducts of normal cellular metabolism can contribute
Ex:
•8-oxo-guanine (oxoG), OH radical attack on guanine; Can lead to mutations
•5-hydroxymethyluracil (HmU), OH radical attack on thymine; Can lead to alterations in protein binding to DNA
• Neutrophils undergo oxidative burst during attack on pathogens (bacteria)
• Generate OH radical and HOCl (hypochlorous acid), which are good for killing outsiders (bacteria), but bad for reacting with self
Ionizing radiation in DNA damage
Direct effect: DNA cleavage
Indirect effect: ROS generation
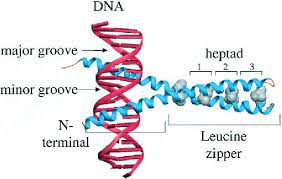 Leucine zipper proteins
Leucine zipper proteins
Leucine zipper proteins are a type of DNA-binding proteins that play a crucial role in gene regulation. They contain a characteristic structural motif called a leucine zipper, which consists of a repeating pattern of leucine residues. This motif allows the proteins to form dimers, where two protein chains come together to bind to specific DNA sequences.
Transcription Factor AP-1
• Very ubiquitous transcription factor
• Stimulated in vivo by a plethora of signals
• Recognizes four thymine methyls for binding
Both HmU and FoU, in the context of base-pairing with A, require repair (Why)
They both have potential to inhibit transcription factor binding
Endogenous Alkylation damage: Damage from endogenous methyl groups - alkylation
• S-adenosylmethionine (SAM or SAMe) – important methyl-donor in normal cellular processes
• If this molecule aberrantly methylates DNA, however, can lead to damage
Ex:
O6-methylguanine (6-meG)
• Changes the H-bonding face, leads to miscoding (with T); Needs repair
N7-methylguanine (7-meG)
• Changes the strength of the glycosidic bond, makes depurination ~1000-fold more likely, leads to abasic site; Needs repair
Endogenous damage by polymerase errors: Damage from polymerase incorporation errors - mismatch
• Sometimes polymerase makes errors
May insert a C opposite an A, or T opposite a G:
• This will lead to a mispair and distortion of the helix
• If mistake is not caught by exonuclease activity, will persist and needs repair
Damage from polymerase incorporation of uracil
• dUTP is found at a certain amount in the nucleotide pool
• Will occasionally be incorporated by polymerase opposite A
• There is no distortion and no mispair here! U:A is Watson-Crick geometry
• However, needs repair because the methyl group of thymine is important for contacts with DNA-binding proteins in the major groove of DNA
 Exogenous DNA damage - UV
Exogenous DNA damage - UV
Damage from sunlight- UV damage
Two major types:
• CPD (cyclobutane pyrimidine dimer)
• 6-4 PP (6-4 photoproduct)
• Both are “thymine dimers”
• Both distort the helix significantly and need repair
•Dimers can also form between an adjacent thymine and cytosine
Exogenous DNA damage: Radiation
Damage from ionizing radiation – oxidation and strand breaks
• SS and DS breaks are very different
• Implications are different
• Repair is different
Damage from environmental toxins: Polycyclic aromatic hydrocarbons – cigarette smoke
• Benzo[a]pyrene derivatives can attach to bases (e.g. addition to guanine);
• This is a “bulky adduct” and needs repair
Damage from environmental toxins: Chemotherapy agents (e.g. bifunctional alkylating agents,
cis-Platin, MMC)
• These agents can crosslink DNA across strands (interstrand crosslinking)
• Lead to major problems for replication and for repair
• Good if you are killing a cancer cell
• Not so good in the case of collateral damage to other cells
Base excision repair
• Repair of uracil when paired with A or G (U:A or U:G)
• Repair of oxidative damage products such as 8-oxo-guanine
• Repair of alkylation damage products such as 3-methyl-adenine
• Repair of hydrolytic deamination products such as thymine mispaired with guanine (T:G)
•Apurinic/apyrimidinic (AP) endonuclease is an enzyme that is involved in the DNA base excision repair pathway (BER).
• A nick is a discontinuity in a double stranded DNA molecule where there is no phosphodiester bond between adjacent nucleotides of one strand
Chemotherapy and base-excision repair 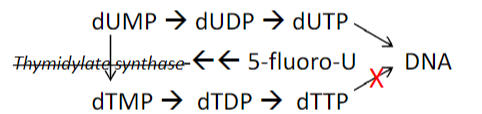 5-fluorouracil is a common chemotherapy agent
5-fluorouracil is a common chemotherapy agent
Toxicity may in part be due to base excision repair!
1. 5-fluorouracil is a suicide inhibitor (inhibits a biological process by becoming covalently bonded to an enzyme) of thymidylate synthase
2. DNA polymerase incorporates U in place of T
3. Repair of uracil will lead to SS breaks repair
4. At a certain threshold, too many strand breaks will trigger apoptosis (futile cycle, thymineless death)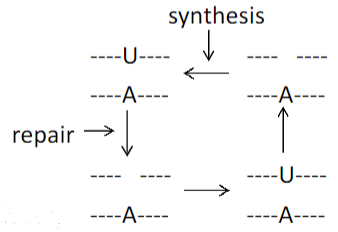
![]()
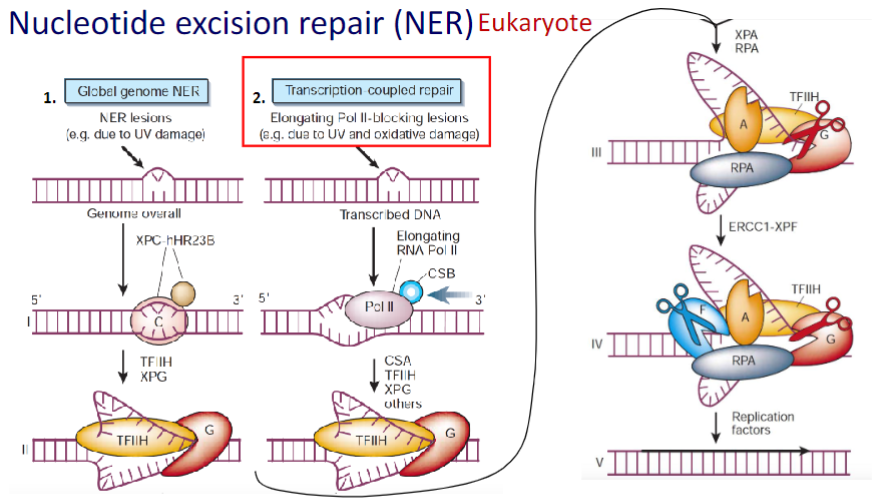
Coupling Nucleotide Excision Repair (NER) to Transcription
- Ensures That the Cell’s Most Important DNA Is Efficiently Repaired
NER repairs the transcribed strands of transcriptionally active genes faster than it repairs non-transcribed strands and transcriptionally silent DNA.
 Mismatch repair in Prokaryote
Mismatch repair in Prokaryote
Mut proteins recognize the mismatch, identify the methylated (parental) strand, and cleave the daughter strand
Segment of daughter strand is released
Polymerase fills the gap, and ligase joins the newly synthesized DNA piece to the original DNA strand
Mismatch repair in Prokaryote MMR deals with correcting mistakes
made by polymerases misincorporatingMMR is of course more complicated in mammalian cells, and the exact mechanism is not yet clearly known about how the daughter strand (with the correct base on it) is identified.
Strand-directed mismatch repair in eukaryotes
The two faces of the clamp differ, and the clamp loader always load the clamp in the same orientation.
With respect to the 3’ end of the previously synthesized Okazaki fragment.
Because all the clamps on the DNA ‘face” in the same direction relative to the replication process, the oriented clamps can be used by the mismatch repair machinery to distinguish newly synthesized DNA from parent DNA.
How strand discrimination occurs on the leading strand is not known with certainty, but it may be similar to what occurs on the lagging strand
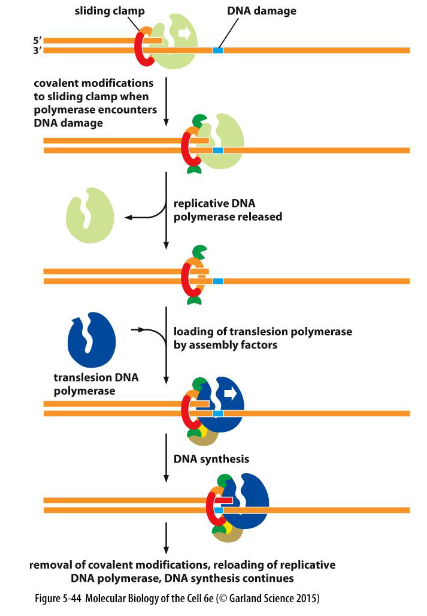
![]() Translesion: Replicative DNA polymerase works together with damaged DNA, which attracts a translesion polymerase specific to that type of damage
Translesion: Replicative DNA polymerase works together with damaged DNA, which attracts a translesion polymerase specific to that type of damage
To avoid the deleterious consequence of a stalled replication fork, cells use specialized polymerases to traverse the damage. This process, termed “translesion DNA synthesis” (TLS)
a replicative polymerase stalled at a site of DNA damage is recognized by the cell as needing rescue.
Once the damaged DNA is bypassed, the covalent modification of the clamp is removed, the translesion polymerase dissociates, and the replicative polymerase is brought back into play
the low-fidelity TLS polymerases also gain access to undamaged DNA where their inaccurate synthesis may actually be beneficial for genetic diversity.
 Direct base repair: used when DNA damage can be chemically reversed
Direct base repair: used when DNA damage can be chemically reversed
Damaged base (alkylated guanine at O-6 position)
Alkyltransferase
- Transfers alkyl group from oxygen of guanine to a cysteine residue in its active site
- MGMT becomes inactivated, ubiquitinated, taken to proteasome for degradation
- Thus, repair of this damage relies on the amount of MGMT around, or ability to synthesize more
Double-strand break repair:
DSB’s can’t be repaired by using the opposite strand
Two major pathways:
End joining (nonhomologous), low fidelity
Homologous recombination, high fidelity
End joining - nonhomologous:
low fidelity or error prone (potentially mutagenic)
ligating two broken ends
used if harder to find homologous chromosome
preferred if you want to generate some errors (VDJ recombination in antibody generation)
![]() Homologous recombination:
Homologous recombination:
High fidelity or error-free
Involves cutting back 5’-ends on both strands, searching for homologous chromosome, strand invasion, DNA synthesis, ligation, and resolving the two DNA strands before moving on
Used when access to homologous chromosome (S and G2)
 BRCA1 and BRCA2 in DNA Double-strand Break Repair
BRCA1 and BRCA2 in DNA Double-strand Break Repair
BRCA1 is a human tumor suppressor gene(caretaker gene)
responsible for repairing DNa
BRCA1 and BRCA2 are unrelated proteins, but both are normally expressed in the cells of breast and other tissue, where they help repair damaged DNA, or destroy cells if DNA cannot
be repaired.A woman’s lifetime risk of developing breast and/or ovarian cancer is greatly increased if she inherits a harmful mutation in BRCA1 or BRCA2.
![]()
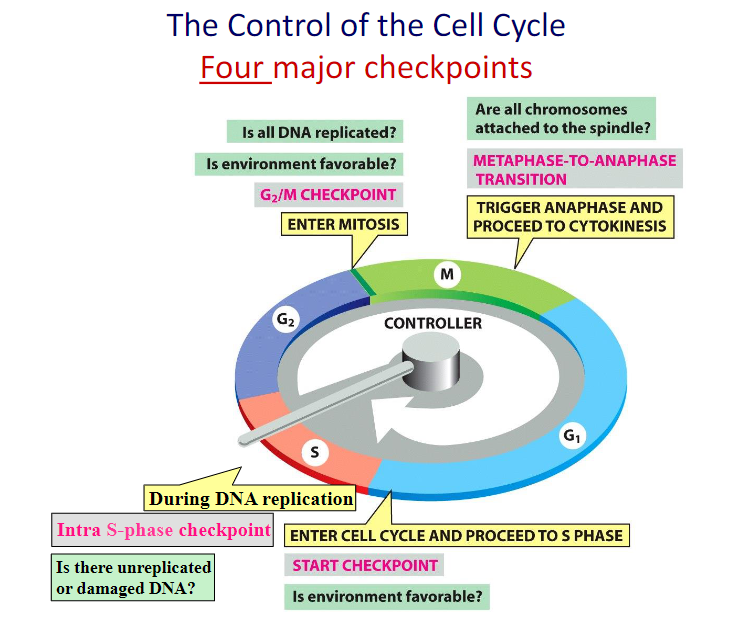
DNA Damage Delays Progression of the Cell Cycle
Because of the importance of maintaining intact, undamaged DNA from generation to generation, eukaryotic cells have an additional mechanism that maximizes the effectiveness of their DNA repair enzymes: they delay progression of the cell cycle until DNA repair is complete.
Thus, in mammalian cells, the presence of DNA damage can block entry from G1 into S phase, it can slow S phase once it has begun, and it can block the transition from G2 phase to M phase.
These delays facilitate DNA repair by providing the time needed for the repair to reach completion. DNA damage also results in an increased synthesis of some DNA repair enzymes.

Soto 5 10/17 DNA Recombination
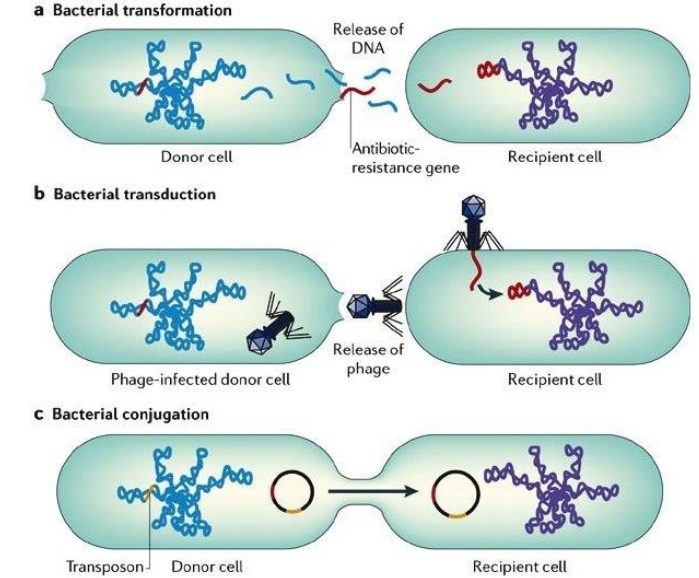
Gene Transfer Mechanism in Bacteria:
Transformation: Transfer of cell-free or “naked” DNA from one cell to another
Transduction: Transfer of genes from one cell to another by a bacteriophage
Conjugation: Transfer of genes between cells that are in physical contact with one another
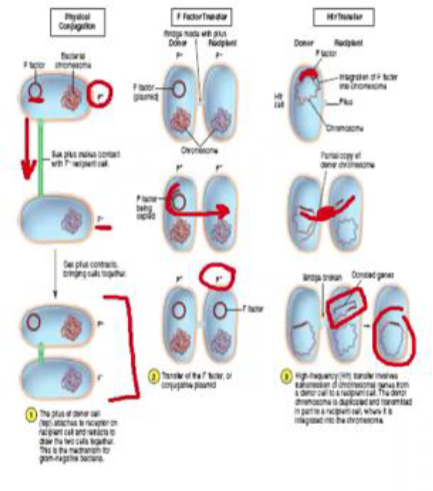
Conjugation: Bacterial “sex”
genetic info is transferred from donor to recipient directly
conjugative pilus
F’ factor (F+/F-)
F Factor transfer
F+ cell makes copy of F factor and transmits it to F-
F- becomes F+
Hfr- High frequency recombination
F plasmid becomes integrated into the F+ chromosome
genes of donor can become integrated into recipient at high frequency
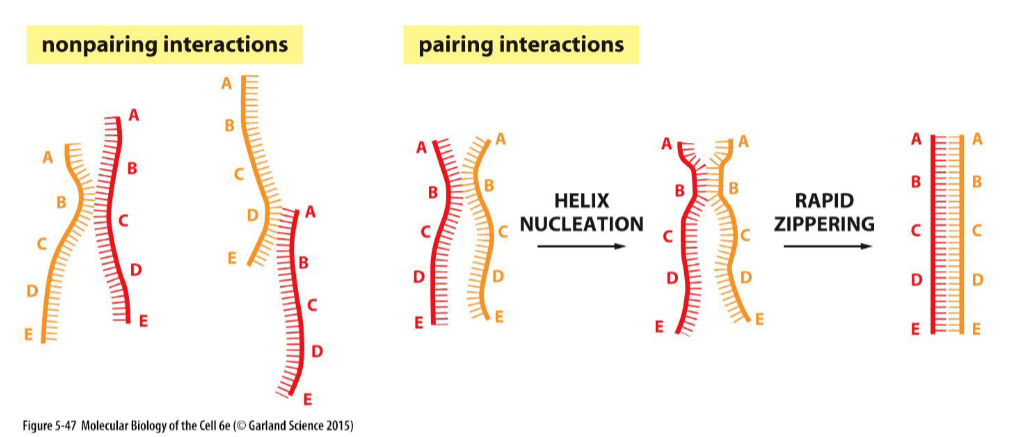
Homologous recombination:
DNA Base-Pairing Guides Homologous Recombination
The vast majority collisions are not productive, as shown on the left, but a few result in a short region where complementary base pairs have formed (helix nucleation).
A rapid zippering then leads to the formation of a complete double helix. Through this trial-and-error process, a DNA strand will find its complementary partner even in the midst of millions of nonmatching DNA strands.
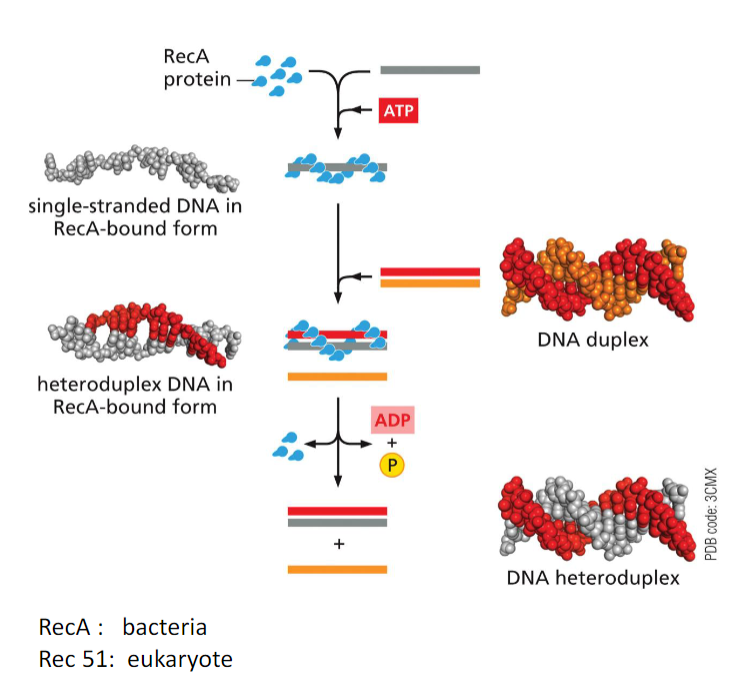
Strand Exchange Is Carried Out by the RecA/Rad51 Protein:
ATP- bound RecA associates with single-strand DNA, holding it in an elongated form where groups of three bases are separated from each other by a stretched and twisted backbone.
In the next step, the RecA- bound single strand then binds to duplex DNA, destabilizing it and
allowing the single strand to sample its sequence through base-pairing, three bases at a time.If no match is found, the RecA-bound single strand of DNA rapidly dissociates and begins
a new search.If an extensive match is found, the structure is disassembled through ATP hydrolysis, resulting in the dissociation of RecA and the exchange of one single strand of DNA for another, thereby forming a heteroduplex.
Homologous Recombination Can Rescue Broken and Stalled DNA Replication Forks:
When a moving replication fork encounters a single-strand break, it will collapse but can be
repaired by homologous recombination.
This pathway allows the fork to
move past the site that was nicked on the original template by using the undamaged duplex as a template to synthesize DNA
![]()
Repair of a stalled replication fork by “fork reversal.”
This mechanism is brought into play when a replication fork stalls when it encounters certain types of damaged nucleotides.
A specialized helicase (not shown) peels the newly synthesized DNA strands away from their parent templates, allowing them to form complementary base pairs with each other and backing up the replication fork.
At this point two outcomes are possible. In the first, because the damaged DNA has been exposed, it can be repaired by conventional repair mechanisms, and the fork can be restarted.
In the second, as shown, DNA synthesis can bypass the damage using newly
synthesized daughter DNA (rather than the damaged parent strand) as the template.This scheme allows the replication fork to move through the DNA damage, which can be repaired at a later time.
Although the initial steps of replication fork reversal are well understood, exactly how the fork restarts afterward remains a mystery.
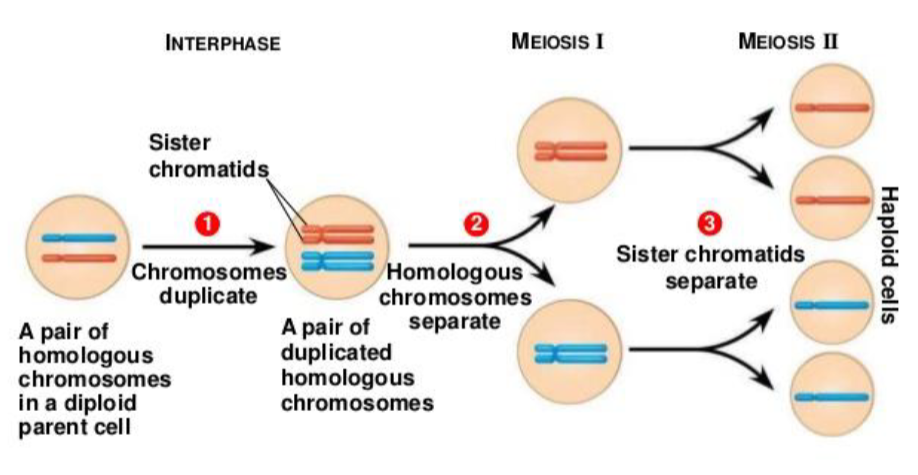
Homologous Recombination Is Crucial for Meiosis:
In animals, plants, fungi
cell division that reduces the chromosome number by half, creating four haploid cells, each genetically distinct from the parent cell that gave rise to them
Meiotic cell divisions are an essential process during oogenesis and spermatogenesis
Although the process of meiosis is related to the more general cell division process of mitosis, it differs in two important respects:
a) Recombination:
- Meiosis: Recombine the genes between the two chromosomes in each pair (one received from each parent), producing recombinant chromosomes with unique genetic combinations in every gamete
- Mitosis: occurs only if needed to repair DNA damage; usually occurs between identical sister chromatids and does not result in genetic changes
b) Chromosome number (ploidy):
- Meiosis: produces four genetically unique cells, each with half the number of chromosomes as in the parent.
- Mitosis: produces two genetically identical cells, each with the same
number of chromosomes as in the parent
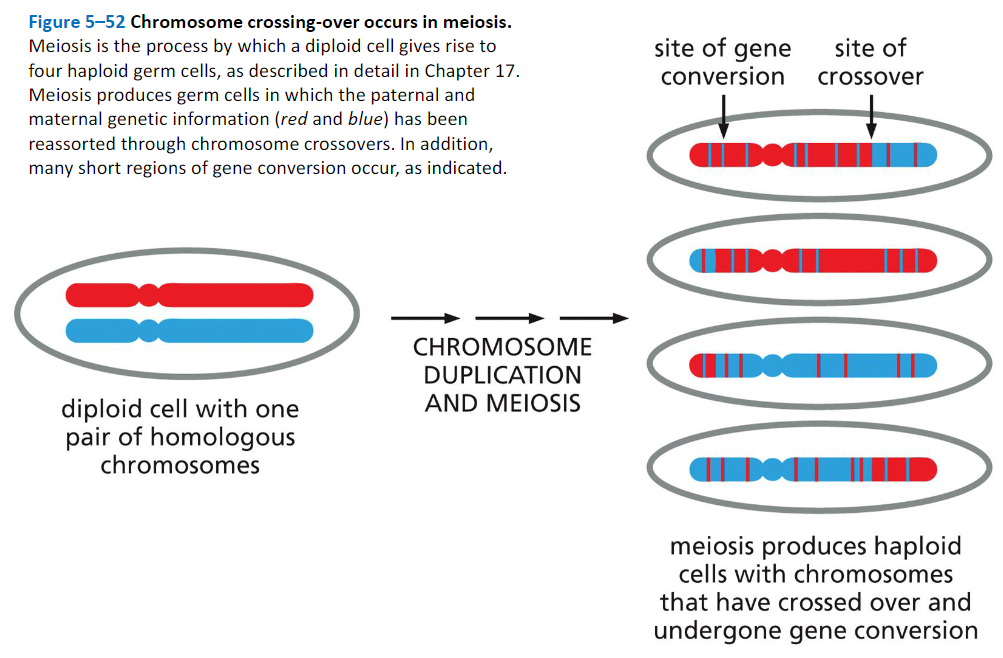
Homologous Recombination Produces Both Crossovers and Non-Crossovers During Meiosis:
Heteroduplexes formed during meiosis.
Heteroduplex DNA is present at sites of recombination that are resolved either as crossovers or non-crossovers. Because the DNA sequences of maternal and paternal chromosomes differ at many positions along their lengths, heteroduplexes often contain a small number of base- pair mismatches, and they are therefore potential sites of gene conversion.
Alternative versions of the same gene are called alleles, and it is the divergence from their expected distribution during meiosis that is known as gene conversion. Genetic studies show that only small sections of DNA typically undergo gene conversion, and in many cases only a part of a gene is changed
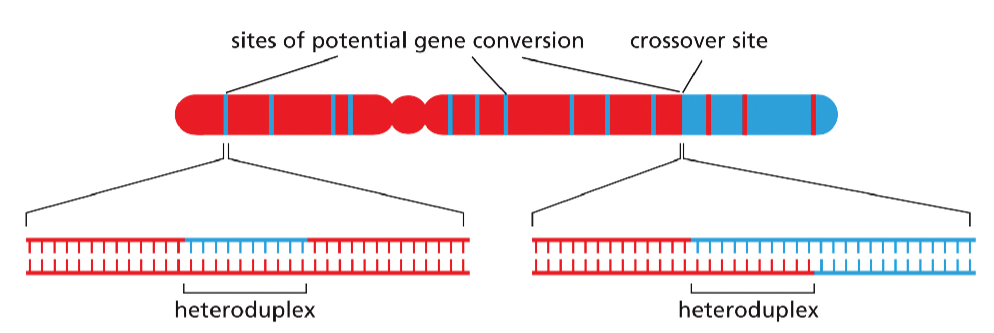
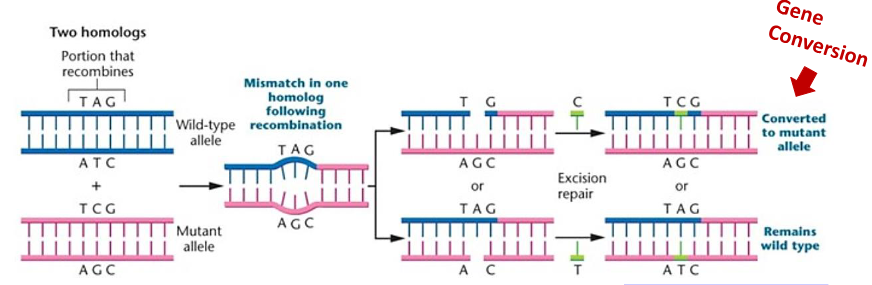
Gene conversion caused by mismatch correction.
In this process, heteroduplex DNA is formed at the sites of homologous recombination between maternal and paternal chromosomes. If the maternal and paternal DNA sequences are slightly different, the heteroduplex region will include some mismatched base pairs, which may then be corrected by the DNA mismatch repair machinery. Such repair can “erase” nucleotide sequences on either the paternal or the maternal strand. The consequence of this mismatch repair is gene conversion, detected as a deviation from the segregation of equal copies of maternal and paternal alleles that normally occurs in meiosis.
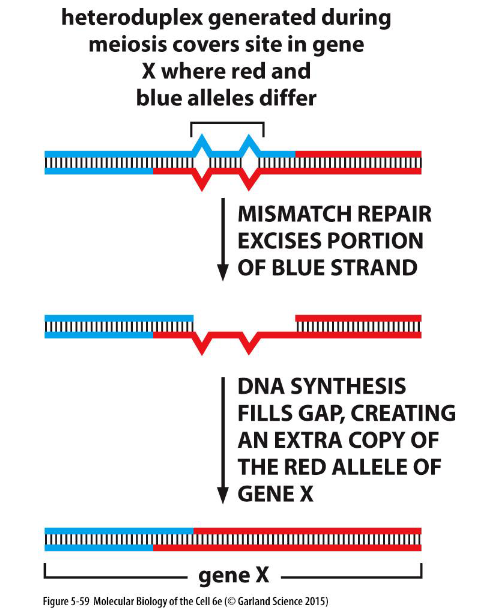
Gene Conversion:
Gene conversion is the process by which one DNA sequence replaces a homologous
sequence such that the sequences become identical after the conversion event. Gene
conversion can be either allelic, meaning that one allele of the same gene replaces another
allele, or ectopic, meaning that one paralogous DNA sequence converts another.Allelic gene conversion occurs during meiosis when homologous recombination between
heterozygotic sites results in a mismatch in base pairing. This mismatch is then recognized
and corrected by the cellular machinery causing one of the alleles to be converted to the
other. This can cause non-Mendelian segregation of alleles in germ cells.
Meiotic Recombination Begins with a Programmed Double-Strand Break:
When the body forms sperm or egg cells in a special type of cell division called meiosis, our DNA mixes and matches in seemingly infinite and unpredictable combinations.
Later, when just two of the great variety of sperm and egg cells meet, they produce children who are different from their parents.
Meiosis would go terribly wrong without crossovers: the swapping of DNA segments between closely aligned pairs of chromosomes, one inherited from each parent
Meiotic crossovers are essential for ensuring correct chromosome segregation
as well as for creating new combinations of alleles for natural selection to take place.
During meiosis, excess meiotic double-strand breaks (DSBs) are generated; a subset of these breaks are repaired to form crossovers, whereas the remainder are repaired as non-crossovers.
What determines where meiotic DSBs are created and whether a crossover or non-crossover will be formed at any particular DSB remains largely unclear
In most eukaryotes, crossovers tend to occur in short, 1–2-kb regions, termed crossover hotspots, where crossover rates can be several magnitudes greater than in the surrounding regions. Interestingly, the distribution and strength of crossover hotspots display substantial
variation among genera, within species, and between sexes.
In some species, the chromosome structure is a major factor shaping crossover landscape because distal euchromatic regions are much more prone to recombination than large heterochromatic centromeric and pericentromeric regions.
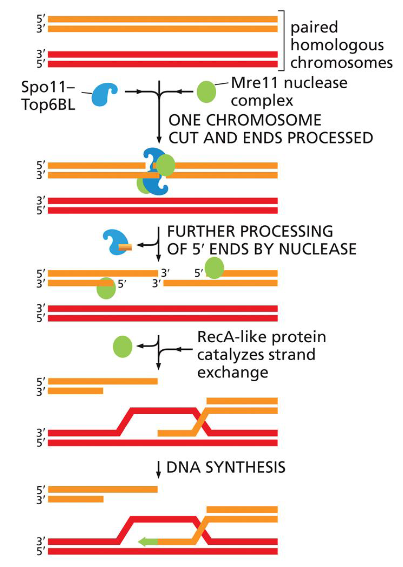
In Meiosis, DNA is intentionally broken!
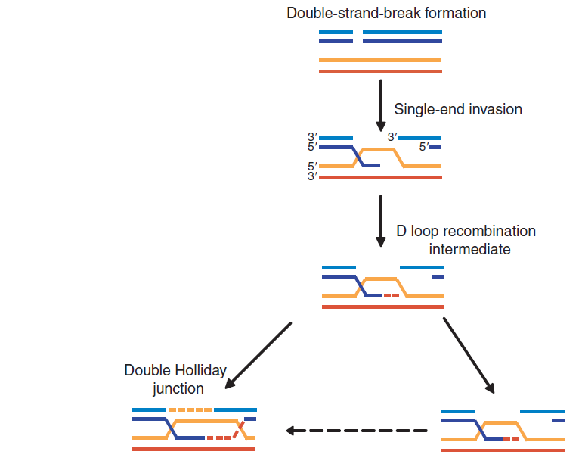
Once
the Spo11 complex (Spo11–Top6BL) and the Mre11 complex break the duplex DNA and process the ends, homologous recombination in meiosis can proceed along alternative pathways.
One (right side of figure) closely resembles the double-strand break repair reaction shown in Figure 5–47 and results in chromosomes that have been “repaired” without crossing over.
The other (left side of figure) proceeds through a double Holliday junction and produces two chromosomes that have crossed over.
During meiosis, both types of homologous recombination take place between the maternal and paternal chromosome homologs because they are held tightly together.
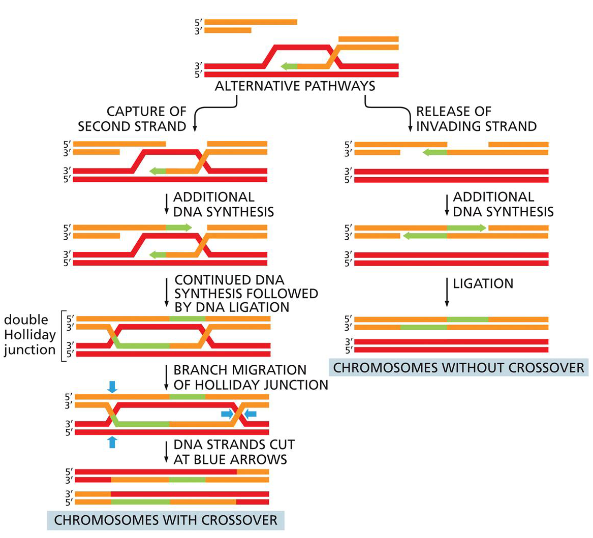
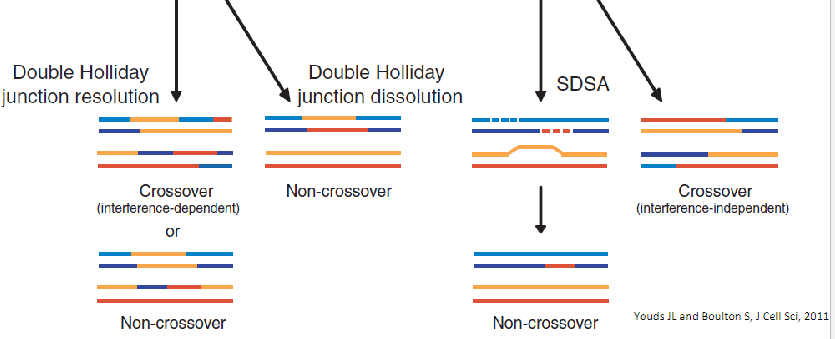
Model for meiotic crossover or non-crossover formation.
Double strand breaks are generated and their 5’ ends are resected to generate a 3’overhang. A strand invasion event then generates a single-end invasion D loop intermediate. If the second end of the original DSB also engages with the homologue, a double Holliday junction is formed (shown on the left). The double Holliday junction can be resolved to form either a crossover (interference-dependent) or a non-
crossover. Alternatively, the junction can be dissolved by double Holliday junction dissolution to form a non- crossover. Instead of forming a double Holliday junction, the D loop can be
dissociated and the invading strand can associate with the opposite end of the original break, as in synthesis- dependent strand annealing (SDSA), to form a non-crossover. Alternatively, the
intermediate can be acted upon by enzymes such as Mus81 that can form interference-independent crossovers.
Through Transposition, Mobile Genetic Elements Can Insert Into Any DNA Sequence:
We have seen that homologous recombination can result in the exchange of DNA sequences between chromosomes. However, the order of genes on the interacting chromosomes typically remains the same following homologous recombination, in as much as the recombining sequences must be very similar for the process to occur.
In this section, we will describe two very different types of recombination—transposition (also called transpositional recombination) and conservative site-specific recombination—that do not require substantial regions of DNA homology.
These two types of recombination reactions can alter gene order along a chromosome and can cause unusual types of mutations that introduce whole blocks of DNA sequence into the genome.
TRANSPOSITION
Transposition and conservative site-specific recombination are largely dedicated to moving a wide variety of specialized segments of DNA— collectively termed mobile genetic elements—from one position in a genome to another. Mobile elements that move by way of transposition are called transposons, or transposable elements. In transposition, a specific enzyme, usually encoded by the transposon itself and typically called a transposase, acts on specific DNA sequences at each end of the transposon, causing it to insert into a new target DNA site.
Most transposons are only modestly selective in choosing their target site, and they can therefore insert themselves into many different locations in a genome. In particular, there is no general requirement for sequence similarity between the ends of the element and the target sequence. Most transposons move only rarely.
In bacteria, where it is possible to measure the frequency accurately, transposons typically move once every 105 cell divisions. More frequent movement would probably destroy the host cell’s genome.
TRANSPOSITION AND CONSERVATIVE SITE-SPECIFIC
RECOMBINATION (1)
• Through Transposition, Mobile Genetic Elements Can Insert Into any DNA Sequence
• DNA-only Transposons Can Move by a Cut-and-Paste Mechanism
• Some DNA-only Transposons Move by Replicating Themselves
• Some Viruses Use a Transposition Mechanism to move
themselves Into Host-Cell Chromosomes
• Some RNA Viruses Replicate and Express Their Genomes Without Using DNA as an Intermediate
• Retroviral-like Retrotransposons Resemble Retroviruses, but
Cannot Move from Cell to Cell
A Large Fraction of the Human Genome Is Composed of Nonretroviral Retrotransposons
• Different Transposable Elements Predominate in Different Organisms
• Genome Sequences Reveal the Approximate Times at Which Transposable Elements Have Moved
• Conservative Site-specific Recombination Can Reversibly Rearrange DNA
• Conservative Site-specific Recombination Can Be Used to Turn Genes On or Off
• Bacterial Conservative Site-specific Recombinases Have
Become Powerful Tools for Cell and Developmental Biologists
TRANSPOSITION AND CONSERVATIVE SITE-SPECIFIC
RECOMBINATION
DNA-Only Transposons Can Move by a Cut-and- Paste Mechanism
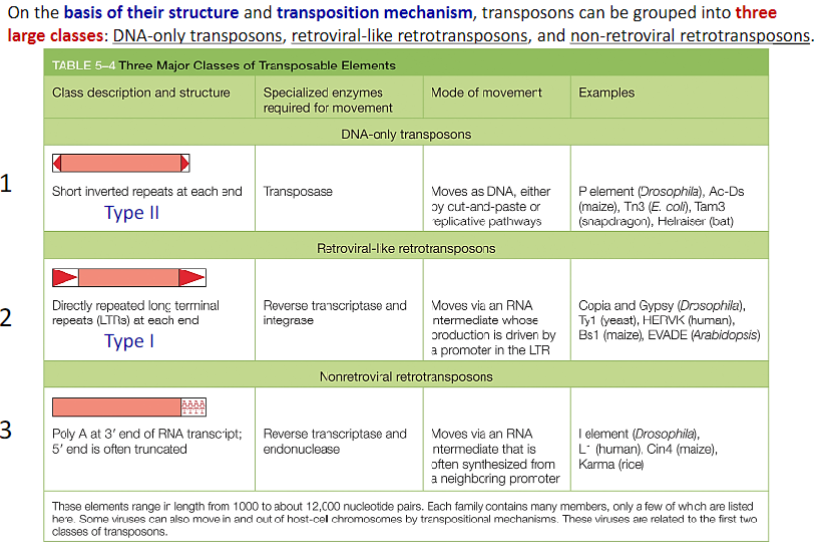
DNA-only transposons, so named because they exist only as DNA during their movement, predominate in bacteria, and they are largely responsible for the spread of antibiotic resistance in bacterial strains.
When antibiotics like penicillin and streptomycin first became widely available in the 1950s, most bacteria that caused human disease were susceptible to them. Now, the situation is different—antibiotics such as penicillin (and its modern derivatives) are no longer effective against many modern bacterial strains.
Transposons often code for the components they need for transposition.
Shown here are two types of bacterial DNA-only transposons. Each carries a gene that encodes a transposase (dark blue and red)— the enzyme that catalyzes the element’s movement—as well as short DNA sequences (light blue and pink) that are recognized by the matching transposase. The short sequences (two in each transposon) are usually arranged so that one is an inverted repeat of the other. Some transposons carry additional genes (yellow) that encode enzymes that inactivate antibiotics such as ampicillin (AmpR). The spread of these transposons is a serious problem in medicine, as it has allowed many disease-causing bacteria to become resistant to the antibiotics developed in the twentieth century.
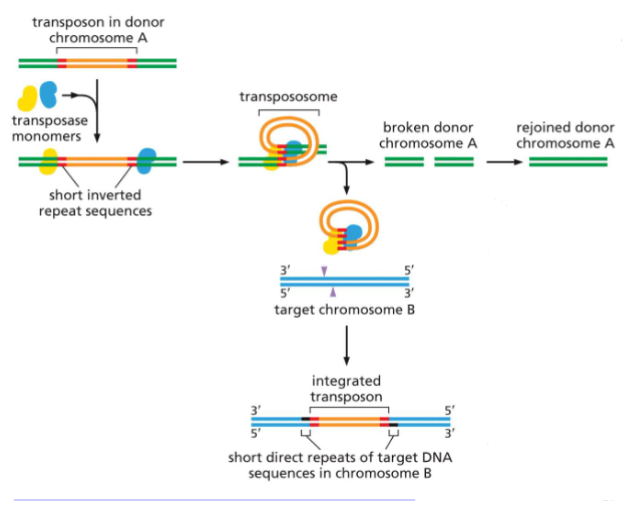
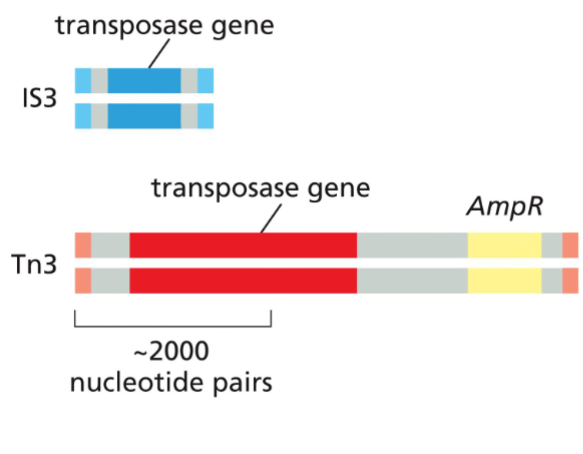 Cut-and-paste transposition.
Cut-and-paste transposition.
DNA-only transposons can be recognized in chromosomes by the inverted repeat DNA sequences (red) present at their ends. These sequences, which can be as short as 20 nucleotides, are all that is necessary for the DNA between them to be transposed by the particular transposase enzyme associated with the element.
The cut-and-paste movement of a DNA-only transposable element from one chromosomal site to another begins when the transposase enzyme brings the two inverted DNA sequences together, forming a DNA loop. Insertion into the target chromosome, also catalyzed by the transposase, occurs at a random site through the creation of staggered breaks in the target chromosome (purple arrowheads).
After the transposition reaction, the single-strand gaps created by the staggered breaks are repaired by DNA polymerase and ligase (black). As a result, the insertion site is marked by a short direct repeat of the target DNA sequence, as shown.
Although the break in the donor chromosome (green) is repaired, this process often alters the DNA sequence, causing a mutation at the original site of the excised transposable element (not shown).
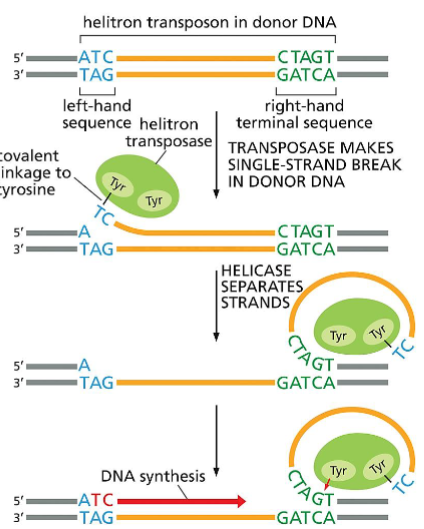
 (Part 1) Mechanism of transposition by helitrons, a type of DNA- only transposon.
(Part 1) Mechanism of transposition by helitrons, a type of DNA- only transposon.
Several models have been proposed for the movement of these recently discovered transposons, and one is shown here. This model is based on studies of a helitron found in bats, called Helraiser. The process begins when the transposase (green) makes a single-strand break at one end of the transposon (blue) and, with the aid of a helicase, “peels back” the single strand.
A second transposase-mediated reaction releases the transposon in the form of single- stranded DNA, which can move to new positions in the host genome. The transposase (which travels with the single- stranded DNA) can then catalyze the covalent insertion of the transposon into a new location in the host DNA.
Transposition by helitrons often moves adjacent host genome sequences along with them. This occurs when, in the third step, the transposase skips over its own CTAGT sequence and cleaves its host DNA downstream at a similar DNA sequence.
According to the model, this skipping produces a single-strand DNA circle that includes both helitron and host DNA, and both are inserted into target DNA.
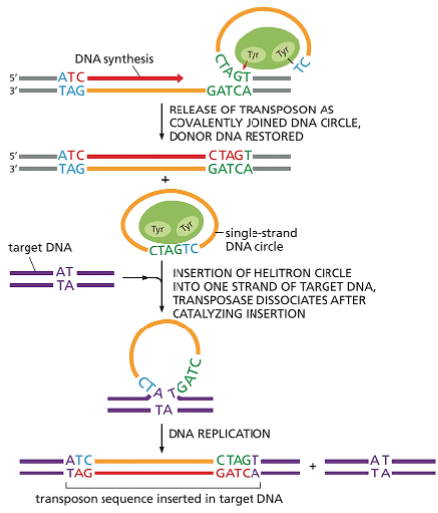
 (Part 2) Mechanism of transposition by helitrons, a type of DNA-only transposon.
(Part 2) Mechanism of transposition by helitrons, a type of DNA-only transposon.
Several models have been proposed for the movement of these recently discovered transposons, and one is shown here. This model is based on studies of a helitron found in bats, called Helraiser.
The process begins when the transposase (green) makes a single-strand break at one end of the transposon (blue) and, with the aid of a helicase, “peels back” the single strand. A second transposase-mediated reaction releases the transposon in the form of single-stranded DNA, which can move to new positions in the host genome.
The transposase (which travels with the single-stranded DNA) can then catalyze the covalent insertion of the transposon into a new location in the host DNA. Transposition by helitrons often moves adjacent host genome sequences along with them. This occurs when, in the third step, the transposase skips over its own CTAGT sequence and cleaves its host DNA downstream at a similar DNA sequence.
According to the model, this skipping produces a single- strand DNA circle that includes both helitron and host DNA, and both are inserted into target DNA.
Some Viruses Use a Transposition Mechanism to Move Themselves Into Host-Cell Chromosomes
Certain viruses are considered mobile genetic elements because they use transposition mechanisms to integrate their genomes into that of their host cell. However, unlike transposons, these viruses encode proteins that package their genetic information into virus particles that can infect other cells.
Many of the viruses that insert themselves into a host chromosome do so by employing one of the first two mechanisms listed in Table 5–4; namely, by behaving like DNA-only transposons or like retroviral-like retrotransposons. Indeed, much of our knowledge of these mechanisms has come from studies of particular viruses that employ them.
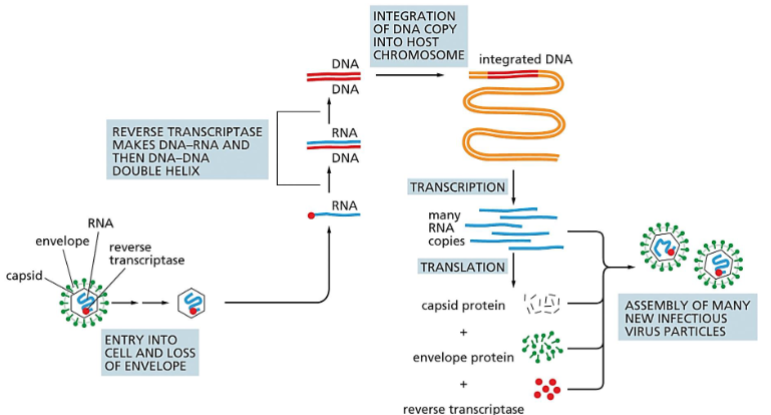
Transposition has a key role in the life cycle of many viruses. Most notable are the retroviruses, which include the human AIDS virus, HIV. Outside the cell, a retrovirus exists as a single-strand RNA genome packed into a protein shell or capsid along with a virus-encoded reverse transcriptase enzyme.
The life cycle of a retrovirus.
The retrovirus genome consists of an RNA molecule (blue) that is typically between 7000 and 12,000 nucleotides in length. It is packaged inside a virus-encoded protein capsid, which is surrounded by a lipid-based envelope that contains virus-encoded envelope proteins (green).
Inside an infected cell, the enzyme reverse transcriptase (red circle) first makes a DNA copy of the viral RNA molecule and then a second DNA strand, generating a double-strand DNA copy of the RNA genome. The integration of this DNA double helix into the host chromosome is then catalyzed by a virus-encoded integrase enzyme.
This integration is required for the synthesis of new viral RNA molecules by the host-cell RNA polymerase, the enzyme that transcribes DNA into RNA (discussed in Chapter 6). As indicated, this viral RNA is then used by host-cell machinery to produce the capsid, envelope, and reverse transcriptase proteins needed to form new virus particles.
Retroviral-like Retrotransposons Resemble Retroviruses, but Lack a Protein Coat
A large family of transposons called retroviral-like retrotransposons move themselves in and out of chromosomes by a mechanism that is similar to that used by retroviruses. These elements are present in organisms as diverse as yeasts, flies, and mammals; unlike viruses, they have no intrinsic ability to leave their resident cell but are passed along to all descendants of that cell through the normal processes of DNA replication and cell division.
A Large Fraction of the Human Genome Is Composed of Non-retroviral Retrotransposons
A significant fraction of many vertebrate chromosomes is made up of repeated DNA sequences. In human chromosomes, these repeats are mostly mutated and truncated versions of non-retroviral retrotransposons, the third major type of transposon. Although most of these transposons in the human genome are immobile, a few retain the ability to move.
Relatively recent movements of the L1 element (sometimes referred to as a LINE or long interspersed nuclear element) have been identified, some of which result in human disease; for example, a particular type of hemophilia results from an L1 insertion into the gene encoding the blood-clotting protein Factor VIII.
Together the LINEs and SINEs make up over 30% of the human genome.
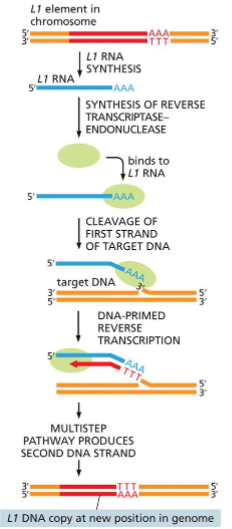
Transposition by a nonretroviral retrotransposon.
Transposition of the L1 element (red) begins when an endonuclease attached to the L1 reverse transcriptase (green) and the L1 RNA (blue) nick the target DNA at the point at which insertion will occur. This cleavage releases a 3ʹ-OH DNA end in the target DNA, which is then used as a primer for the reverse transcription step shown.
This generates a single-strand DNA copy of the element that is directly linked to the target DNA. In subsequent reactions, further processing of the single-strand DNA copy results in the generation of a new double-strand DNA copy of the L1 element that is inserted at the site of the initial nick.
 TRANSPOSITION AND CONSERVATIVE SITE-SPECIFIC RECOMBINATION
TRANSPOSITION AND CONSERVATIVE SITE-SPECIFIC RECOMBINATION
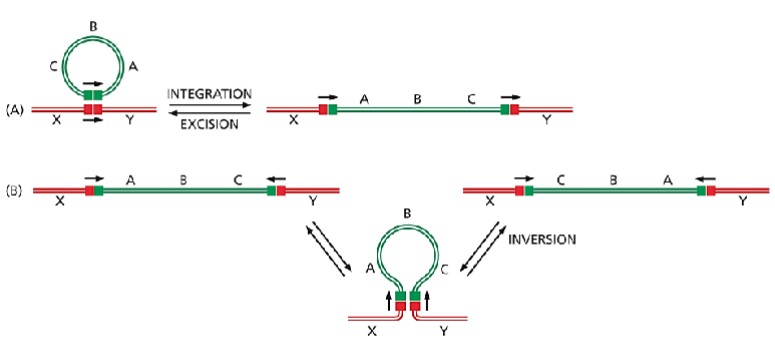
Conservative Site-Specific Recombination Can Reversibly Rearrange DNA
A different kind of recombination mechanism, known as conservative site-specific recombination, rearranges other types of mobile DNA elements. In this pathway, breakage and joining occur at two special sites, one on each participating DNA molecule. Depending on the positions and relative orientations of the two recombination sites,
DNA integration, DNA excision, or DNA inversion can occur. Conservative site-specific recombination is carried out by specialized enzymes that break and rejoin two DNA double helices at specific sequences on each DNA molecule. The same enzyme system that joins two DNA molecules can often take them apart again, precisely restoring the sequence of the two original DNA molecules.
Two types of DNA rearrangement produced by conservative site-specific recombination.
The only difference between the reactions in (A) and (B) is the relative orientation of the two short DNA sites (indicated by arrows) at which a site-specific recombination event occurs. (A) Through an integration reaction, a circular DNA molecule can become incorporated into a second DNA molecule; by the reverse reaction (excision), it can exit to re-form the original DNA circle. Many bacterial viruses move in and out of their host chromosomes in this way. (B) Conservative site-specific recombination can also invert a specific segment of DNA in a chromosome. A well-studied example of DNA inversion through site-specific recombination occurs in the bacterium Salmonella typhimurium, an organism that is a major cause of food poisoning in humans; as described in the following section, the inversion of a DNA segment changes the type of flagellum that is produced by the bacterium
 Conservative Site-Specific Recombination Can Be Used to Turn Genes On or Off
Conservative Site-Specific Recombination Can Be Used to Turn Genes On or Off
Many bacteria use conservative site-specific recombination to control the expression of particular genes. A well-studied example occurs in Salmonella bacteria and is known as phase variation.
The switch in gene expression results from the occasional inversion of a specific 1000-nucleotide-pair piece of DNA, brought about by a conservative site-specific recombinase encoded in the Salmonella genome.
This change alters the expression of the cell-surface protein flagellin, for which the bacterium has two different genes (Figure 5–65). With the promoter in one orientation, the bacteria synthesize one type of flagellin; with the promoter in the other orientation, they synthesize the other type.
The recombination reaction is reversible, allowing bacterial populations to switch back and forth between the two types of flagellin.
Phase variation helps protect the bacterial population against the immune response of its vertebrate host. If the host makes antibodies against one type of flagellin, a few bacteria whose flagellin has been altered by gene inversion will still be able to survive and multiply.
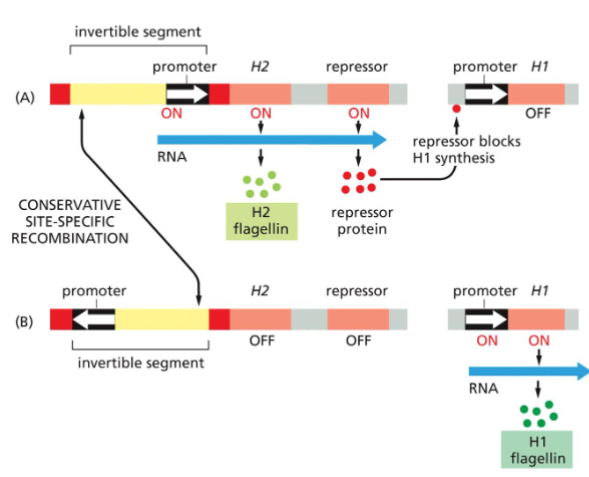
Switching gene expression by DNA inversion in bacteria.
Alternating transcription of two flagellin genes in a Salmonella bacterium is caused by a conservative site-specific recombination event that inverts a small DNA segment containing a promoter.
(A) In one orientation, the promoter activates transcription of the H2 flagellin gene as well as that of a repressor protein that blocks the expression of the H1 flagellin gene.
Promoters and repressors are described in detail in Chapter 7; here we note simply that a promoter is needed to express a gene into protein and that a repressor blocks this from happening.
(B) When the promoter is inverted, it no longer turns on H2 or the repressor, and the H1 gene, which is thereby released from repression, is expressed instead. The inversion reaction requires specific DNA sequences (red) and a recombinase enzyme that is encoded in the invertible DNA segment.
This site-specific recombination mechanism is activated only rarely (about once in every 105 cell divisions). Therefore, the production of one or the other flagellin tends to be faithfully inherited in each clone of cells.
Bacterial Conservative Site-Specific Recombinases Have Become Powerful Tools for Cell and Developmental Biologists
Like many of the mechanisms used by cells and viruses, site-specific recombination has been put to work by scientists to study a wide variety of problems.
To decipher the roles of specific genes and proteins in complex multicellular organisms, genetic engineering techniques are used to produce worms, flies, and mice carrying a gene encoding a site-specific recombination enzyme plus a carefully designed target DNA with the DNA sites that this enzyme recognizes.
At an appropriate time, the gene encoding the enzyme can be activated to rearrange the target DNA sequence. Such a rearrangement is widely used to delete a specific gene in a particular tissue of a multicellular organism (Figure 5–66)
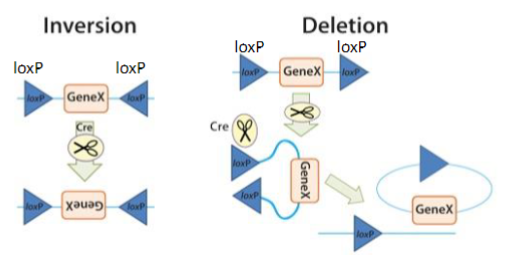
Cre-lox Recombinase System
The Cre-lox system is a technology that can be used to induce site-specific recombination events. The system consists of two components derived from the P1 bacteriophage: the Cre recombinase and a loxP recognition site.
The P1 bacteriophage uses these components as part of its natural viral lifecycle, and researchers have adapted the components for use in genome manipulation. Cre recombinase, originally named because it “causes recombination”. LoxP (locus of X(cross)-over in P1) sites are 34-base-pair long recognition sequences consisting of two 13-bp long palindromic repeats separated by an 8-bp long asymmetric core spacer sequence.
The asymmetry in the core sequence gives the loxP site directionality, and the canonical loxP sequence is ATAACTTCGTATA-GCATACAT-TATACGAAGTTAT. The loxP sequence does not occur naturally in any known genome other than P1 phage, and is long enough that there is virtually no chance of it occurring randomly. Therefore, inserting loxP sites at deliberate locations in a DNA sequence allows for very specific manipulations.
As alluded to above, the Cre recombinase catalyzes the site specific recombination event between two loxP sites, which can be located either on the same or on separate pieces of DNA. Both 13bp repeat sequences on a single loxP site are recognized and bound by a Cre protein, forming a dimer. The two loxP sites then align in a parallel orientation, allowing the four Cre proteins to form a tetramer. A double-strand DNA break occurs within the core spacer of each loxP site and the two strands are ligated, resulting in the reciprocal crossover event.
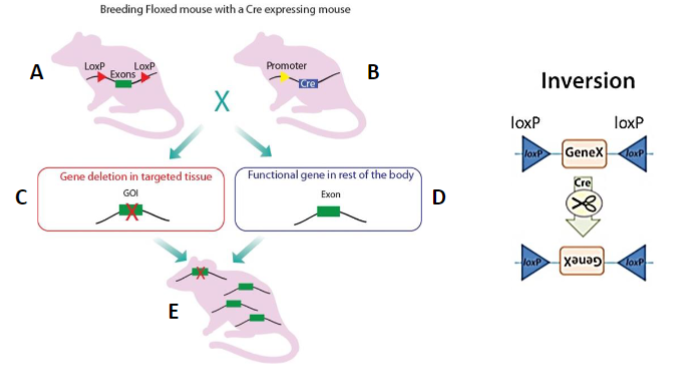
Inversion: If the loxP sites are on the same DNA strand and are in opposite orientations, recombination results in an inversion and the region of DNA between the loxP sites is reversed.
Deletion: If the sites face in the same direction, the sequence between the loxP sites is excised as a circular piece of DNA (and is not maintained).
 Bacterial Conservative Site-Specific Recombinases Have Become Powerful Tools for Cell and Developmental Biologists
Bacterial Conservative Site-Specific Recombinases Have Become Powerful Tools for Cell and Developmental Biologists
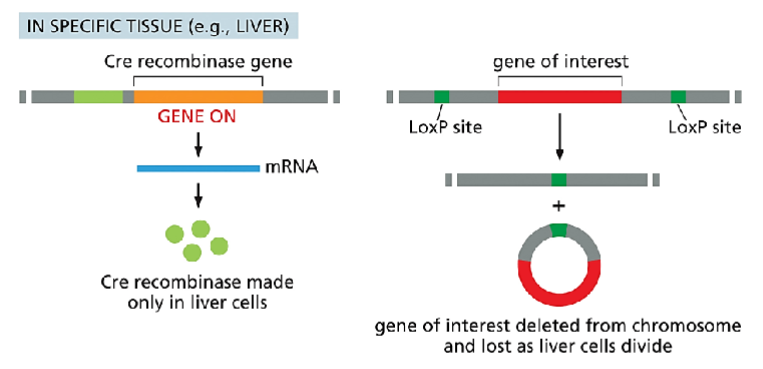
Figure 5–66 (Part 1) How a conservative site-specific recombination enzyme from bacteria is used to delete a specific gene from a particular mouse tissue. This approach requires the insertion of two specially engineered DNA molecules into the animal’s germ line.
The first contains the gene for a recombinase (in this case, the Cre recombinase from the bacteriophage P1) under the control of a tissue-specific promoter that ensures the recombinase is expressed only in that tissue.
The second DNA molecule contains the gene of interest, flanked by the DNA sequences of the recognition sites for the recombinase (in this case, LoxP sites). The mouse has been engineered to contain only this copy of the gene of interest.
Therefore, if the recombinase is expressed only in the liver, the gene of interest will be deleted there, and only there. As described in Chapter 7, many tissue-specific promoters are known; moreover, many of these promoters are active only at specific times in development.
Thus, this method makes it possible to study the effects of deleting any gene of interest at specific times during the development of each tissue. For this reason, it is a powerful tool for scientists investigating the role of individual genes in animal and plant development.
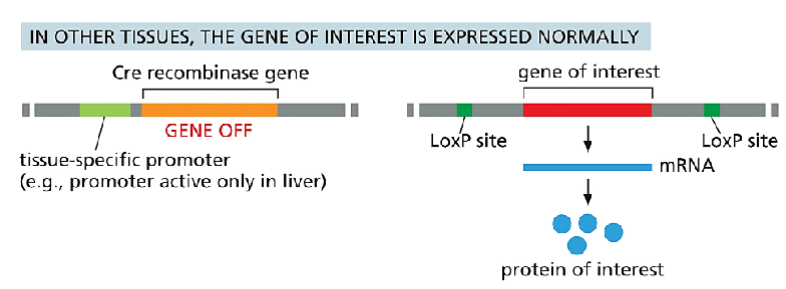
Part 2) How a conservative site-specific recombination enzyme from bacteria is used to delete a specific gene from a particular mouse tissue.
This approach requires the insertion of two specially engineered DNA molecules into the animal’s germ line. The first contains the gene for a recombinase (in this case, the Cre recombinase from the bacteriophage P1) under the control of a tissue-specific promoter that ensures the recombinase is expressed only in that tissue.
The second DNA molecule contains the gene of interest, flanked by the DNA sequences of the recognition sites for the recombinase (in this case, LoxP sites). The mouse has been engineered to contain only this copy of the gene of interest.
Therefore, if the recombinase is expressed only in the liver, the gene of interest will be deleted there, and only there. As described in Chapter 7, many tissue-specific promoters are known; moreover, many of these promoters are active only at specific times in development.
Thus, this method makes it possible to study the effects of deleting any gene of interest at specific times during the development of each tissue. For this reason, it is a powerful tool for scientists investigating the role of individual genes in animal and plant development.
Cre-Lox system is used as a bioengineering technique
Crossbreeding the floxed mouse with a Cre-recombinase expressing mouse produces a condition knockout model. The Cre expression is driven by a promoter of choice (yellow triangle, a tissue-specific or ubiquitous promoter). Expressed Cre recombinase deletes the floxed exon(s) spatial-specifically, thereby causing a frame shift in the downstream sequence.
Lecture 6 Soto DNA-RNA Purif-EMSA-ChiP
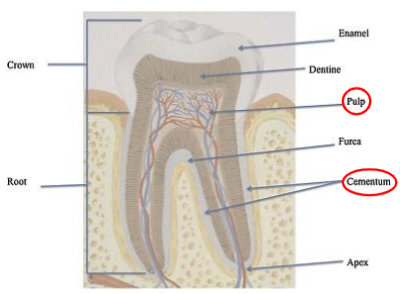
DNA in teeth
pulp and cementum are a source of mtDNA
enamel preserves dentine and pulp
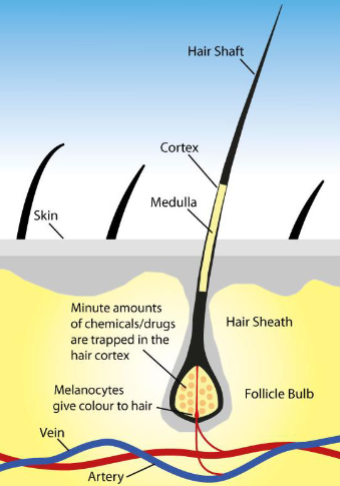
 DNA in hair
DNA in hair
There is no nuclear DNA present in the hair shaft
Hair root or follicle contains DNA
Not used for DNA testing
 Collecting DNA samples for a DNA test
Collecting DNA samples for a DNA test
DNA testing is commonly done with the use of an oral swab
gentle friction from the swab collects epithelial membrane cells to attach
DNA into Red blood cells (RBC)
Human RBC do not contain DNA
RBC from birds or some other species do contain DNA
Old method of DNA purification:
DNA purification: plasmid purification
grow colony on plate
pick single colony
grow in culture in Erlenmeyer flask
Lyse cells
wash and elute in columns
Product: purified plasmid DNA
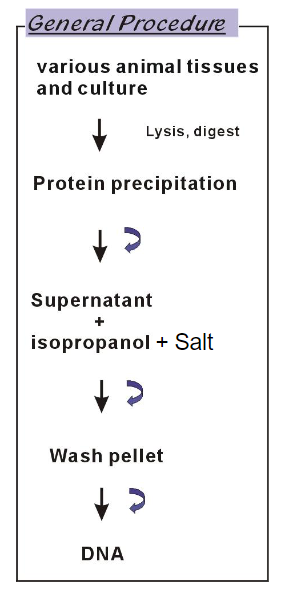
DNA purification: Animal tissue or cell culture purification
lysis, digest
Bind to column
wash, elute
Product: purified DNA
The columns contain silica resin that binds to DNA and RNA, depending on the kit
RNase free DNase: acts on RNA, has no DNA
DNase free RNase acts on DNA, has no RNA
The first step in RNA and DNA extraction: Lysis
contains high concentration of chaotropic salt
destabilizes hydrogen bonds (and VDW forces and hydrophobic interactions)
Chaotropic salts include guanidine HCL, guanidine thiocyanate, urea, and lithium
perchlorate
Column binding
silica has the ability to attract nucleic acids
needs TE (tris-EDTA) buffer or water to break the attraction between silica and nucleic acids
RNA purification (toxic/long way)
Several RNases are partially resistant to high temperatures (autoclave) therefore must be inactivated by chemicals (DEPC, Diethylpyrocarbonate)
Animal tissue
homogenized with LN2
Lysis buffer
Chloroform, take supernatant
Phenol/ chloroform, take supernatant
Isopropanol, take supernatant
Wash RNA pellete
Add RNA hydration buffer
A phenol-chloroform extraction is a liquid-liquid extraction. A liquid-liquid extraction is a method that separates mixtures of molecules based on the differential solubilities of the individual molecules in two different immiscible liquids
RNA purification (non-toxic/short way) with columns
Lyse
homogenize
bind
wash
elute
Purified RNA
DNA and RNA have an absorbance at 260 nm (UV light)
DNA analysis:
EthBr intercalates into double strand DNA and produce fluorescence
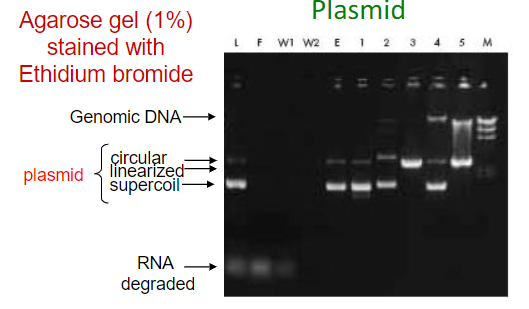
Plasmid: multiple bands for the different segments of the plasmids and a separate band for genomic DNA
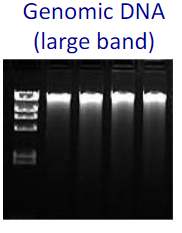 Genomic DNA: Large Bright bands
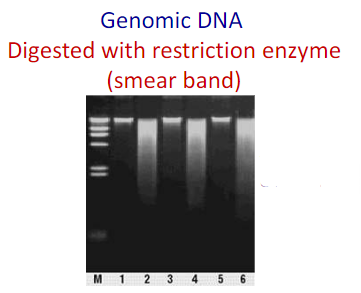
Genomic DNA: Large Bright bands
when digested with restriction enzymes there could be smeared bands
Electrophoretic Mobility Shift Assay (EMSA)
A transcription factor molecule binds to the DNA in a regulatory region upstream of a
gene (promoter region), and thus regulates the production of a protein from a gene
The EMSA technique is based on the observation that protein:DNA complexes
migrate more slowly than free linear DNA fragments when subjected to non-
denaturing polyacrylamide or agarose gel electrophoresis
Protein:DNA complexes formed on linear DNA fragments result in the
characteristic retarded mobility in the gel
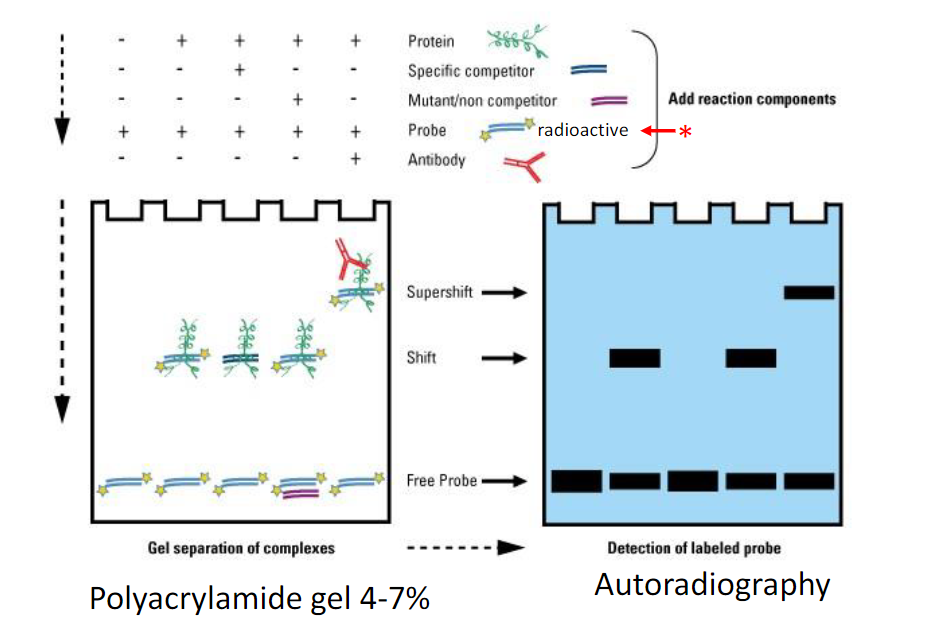
Bottom: free DNA is at the bottom as it runs the fastest when unattached
Middle: DNA +protein attach and run slower than just DNA
Top: Super complexes between DNA, protein, etc. (larger and heavier structure)
Canonical vs non-canonical sequences
Canonical: the presence of the most common nucleotides in a sequence that binds to a transcription factor
the most common choice of base or amino acid at each position
Larger the letter size, higher frequency in specific location
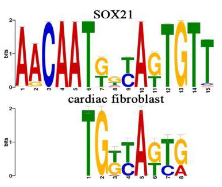 Non- canonical: When researchers find a new feature in an established pathway that does not fit into the canonical model, it is referred to a non-canonical
Non- canonical: When researchers find a new feature in an established pathway that does not fit into the canonical model, it is referred to a non-canonical
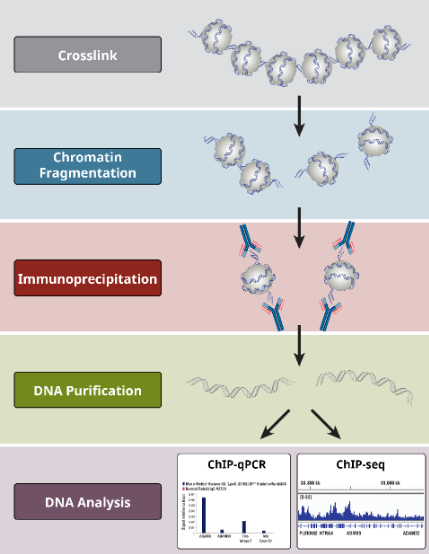
Chromatin immunoprecipitation (ChIP) assay
Formaldehyde: fixation
Restriction enzyme: shorten
Chromatin fragmentation by digestion
Purification: beads
Reversal crosslinking: heat or digestion
Sequence of the isolated DNA: via PCR, qPCR
EMSA vs CHIP
If you just want to show that your proteins are able to bind to the given sequence, you can do a gel shift. If you want to assess whether binding is actually occurring in the cell, you will have to do ChIP. ChIP does requires a large time investment, whereas the gel shift can be done pretty quickly. You should probably do the gel shift first to determine binding, and if necessary, confirm in vitro using chip.
Lec 7 Soto PCR-pPCR-RT-PCR-DNA microarray
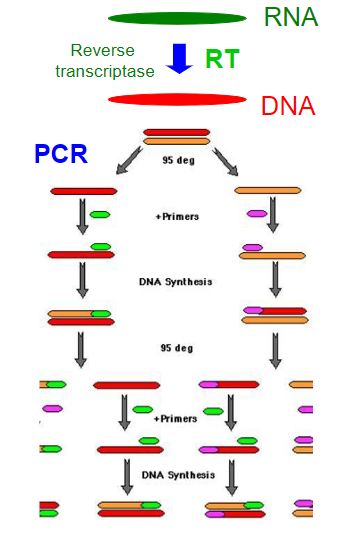
PCR
Polymerase chain reaction, or PCR, is a laboratory technique used to make
multiple copies of a segment of DNA
Heat (95 degree C) separates strands
Binds to DNA primers
Taq polymerase replicates DNA
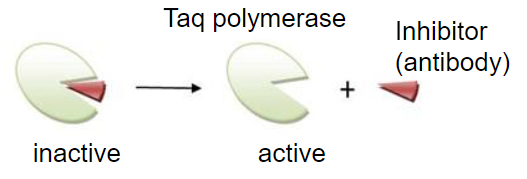
Hot start PCR
inhibit polymerases activity at ambient temp: reduces non-specific priming
High Fidelity PCR
enzyme mix: recombinant Taq DNA polymerase plus DNA polymerase containing proofreading ability by virtue of its 3’ → 5’ exonuclease activity
Increases fidelity 6x
usually hot start system
Special polymerase Pfu does not make ligatable blunt ends
Primers for PCR
Primer melting temperature (Tm): Tm = 2 (A + T) + 4(C + G) degree Celsius
Annealing temperature (Ta): Ta = Tm – 5 degree Celsius
Forward primer copies DNA antisense strand, non-coding strand
Reverse primer copies DNA sense strand, coding strand
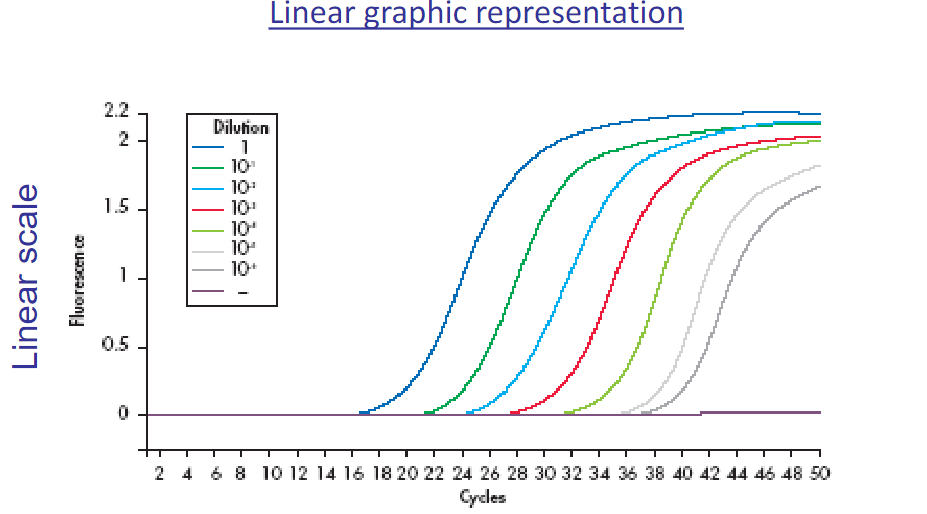
Real-Time (quantitative PCR, qPCR)
fluorescent dyes whose fluorescent signal is proportional to the DNA content in each cycle
allow for the detection of PCR amplification during the early phases of the reaction
Wide dynamic detection range: The range of initial template concentrations over which accurate measurement can be achieved
uses a small amplicon: 75 to 200 bases in length
DNA binding agents: SYBR 1 dye
intercalate with any double strand DNA, non-sequence specific dye
SYBR 1 signal intensities correlate with DNA amplification of the sample
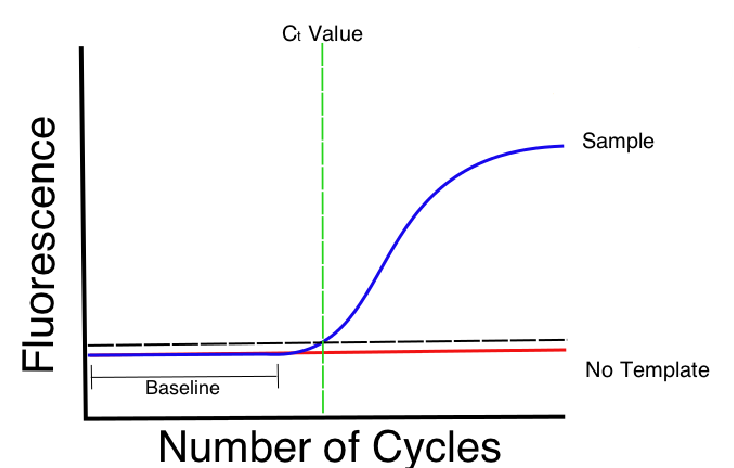
CT value: The spot where your reaction curve intersects your threshold line is the “threshold cycle” (CT)
This spot shows the number of cycles it took to detect a real signal from your samples
ΔCT: In PCR, the delta Ct value is the difference between the Ct value of the target gene and the Ct value of the reference gene (GAPDH).
ΔΔCT: Delta-Delta Ct (ΔΔCt) is a mathematical term that describes the difference between the ∆Ct values of a treated sample and an untreated sample
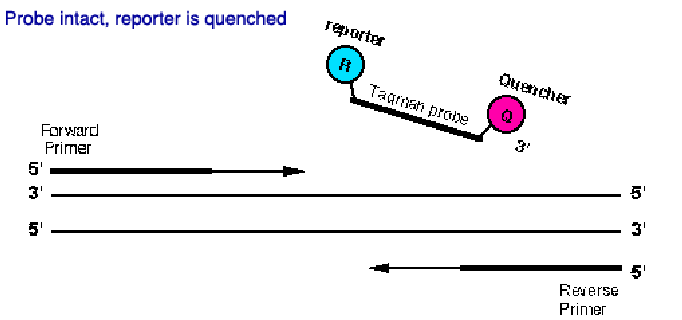
Hydrolysis probes: Taqman probe
sequence-specific DNA fluorescent probes
The TaqMan probe contains a reporter dye at the 5′ end of the probe and a quencher dye at the 3′ end of the probe
During the reaction, cleavage of the probe results in increased florescnence
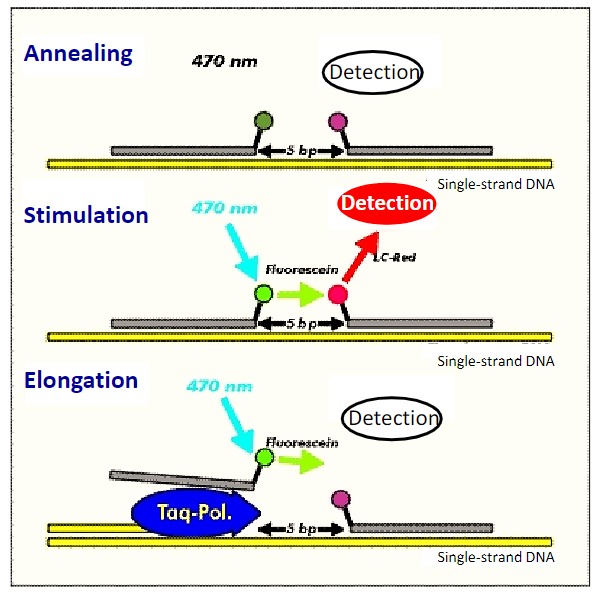
FRET (Fluorescence resonance energy transfer) reaction
(sequence-specific DNA fluorescent probes)
oligonucleotides bind to neighbored on a target nucleic acid
The amount of hybridized probes increases with the growing PCR product
The signal is proportional to the amount of amplicon

Amplicon Selection
The amplicon is the region of target sequence that is to be analyzed and is encompassed by the forward and reverse PCR primers
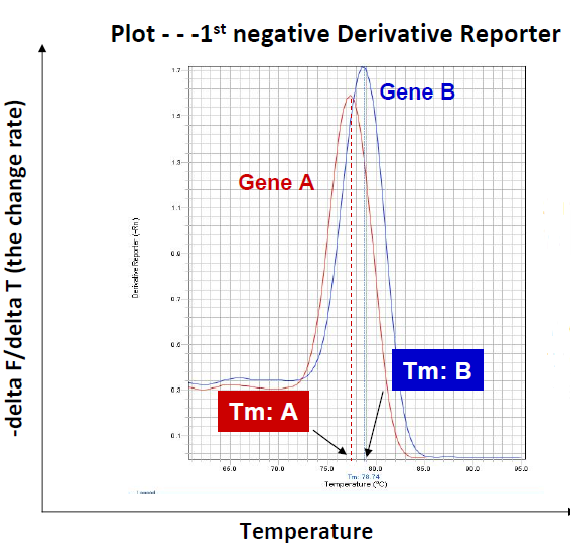
Higher melting temperatures means larger structures
Single melt curve of each amplicon is required for specificity valifdation
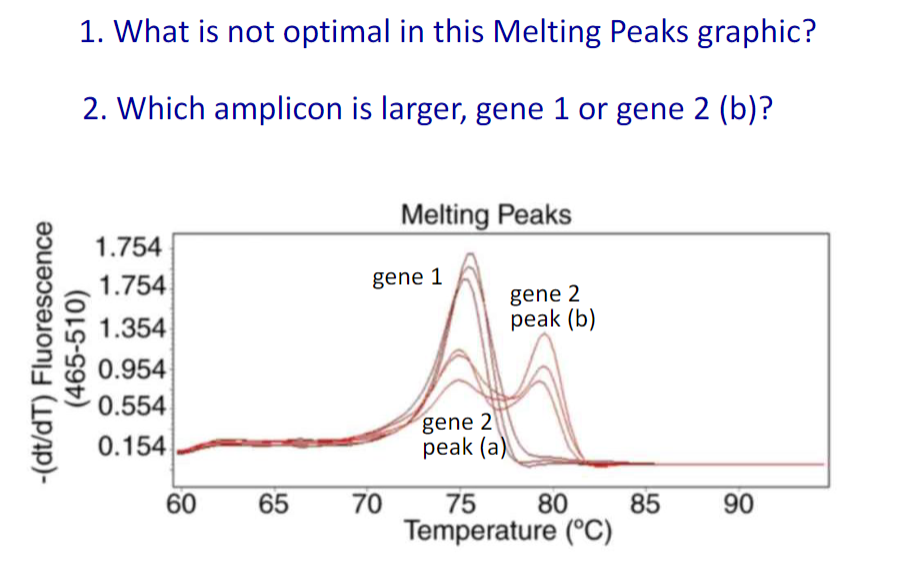
Gene 2 has two different melting peaks
Gene 2
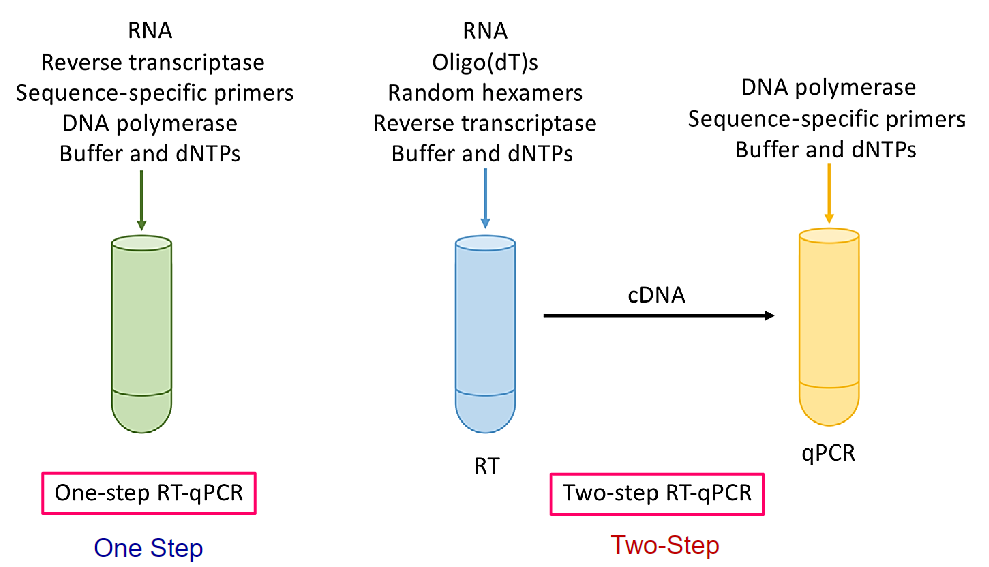
Reverse transcription PCR (RT-PCR)
To perform RT-PCR we need to purify RNA and convert mRNA into DNA (cDNA) by using reverse transcriptase (RT). RT-PCR is useful to measure the expression of a gene.
From mRNA is produced DNA (copy, cDNA) by using reverse transcriptase (RT). This cDNA is amplified by PCR. The size of the amplified DNA differs of the genomic DNA if introns are present in the gene and primers are located in two different exons.
Four basic reagents needed to produce cDNA:
mRNA as template
dNTPs
reverse transcriptase
primers
![]() 1 vs 2 step RT-qPCR
1 vs 2 step RT-qPCR
Lec 8 Soto Restriction enzymes RFLP cloning
A bacterium uses a restriction enzyme to defend against bacterial viruses called bacteriophages, or phages. When a phage infects a bacterium, it inserts its DNA into the bacterial cell so that it might be replicated. The restriction enzyme prevents replication of the phage
DNA by cutting it into many pieces
Restriction enzymes were named for their ability to restrict, or limit, the number of strains of bacteriophage that can infect a bacterium
3 classes of Restriction Endonucleases
class 1 and 3 enzymes cleave the DNA at sites distant from the recognition site
Class 2 enzymes cleave DNA within the recognition sequence
some yield blunt ends: straight cuts that yield no availability for annealing
some yield overhanging (sticky) ends: staggered cut allows for annealing
EcoR1 has a recognition sequence that is a palindrome, leaves 2 sticky ends
Restriction fragments can be separated on the basis of size on a gel, small move quickly while large move slowly
![]()
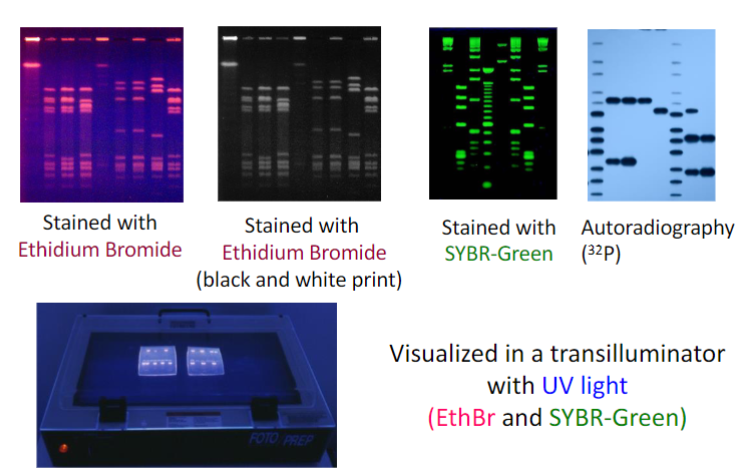 Restriction Fragments can be visualized in a number of ways
Restriction Fragments can be visualized in a number of ways
• Radioactively labeled(32P) polynucleotides can be detected by autoradiography or variants thereof
• DNA fragments can be stained with Ethidium bromide and be
visualized with UV light
• DNA fragments can be labeled with fluorescent markers and seen under UV light
• Specific sequences can be visualized by hybridization with a
radiolabeled probe
 1. B, 2. A, 3. C
1. B, 2. A, 3. C
Southern, Northern, and Western blot
Southern: DNA analysis
Northern: RNA analysis
Western: Protein analysis
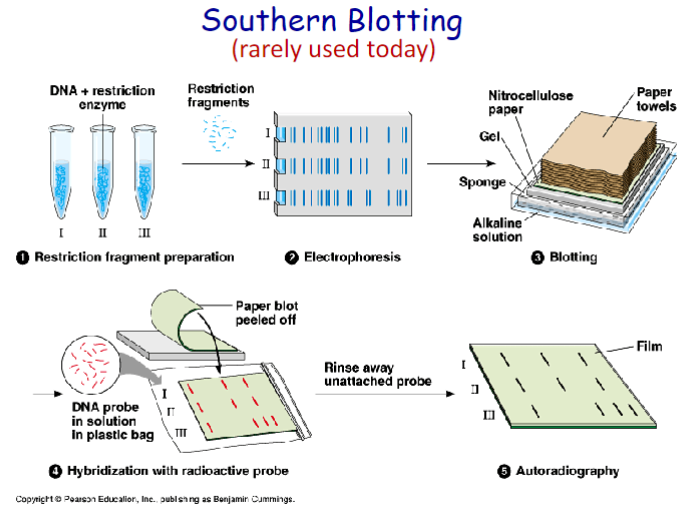
Southern Blot
DNA separated by gel electrophoresis
DNA bonded onto nitrocellulose
specific DNA probe added
on X-ray film the DNA of interest is revealed
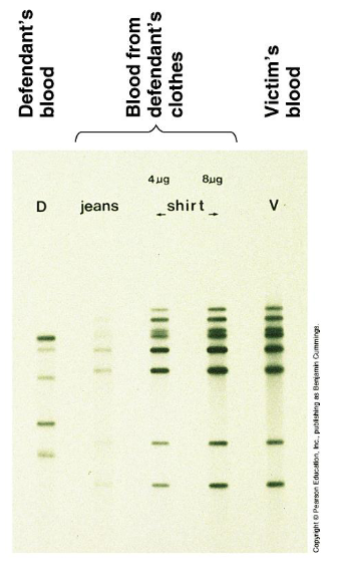
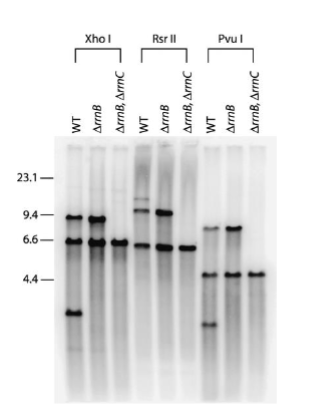 Southern blotting can be used to:
Southern blotting can be used to:
investigate whether a gene is amplified, deleted,
or structurally rearranged in cancer cells as
compared to normal cells
Detect the presence of repetitive sequences, such
as transposable elements, that are present in
multiple copies within the chromosomal DNA of an
organism
Accurately size repeat expansions that are too large to amplify through PCR
Can be used to determine an individual’s unique collection of DNA restriction fragments
Need a probe: A short single stranded DNA which is
complementary to the region of interest
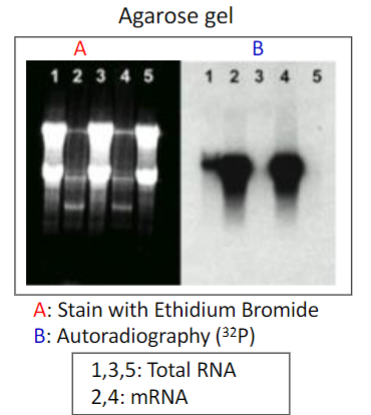
Northern blot:
RNA separation by gel electrophoresis
RNA blot on nitrocellulose
label with specific nucleic acid probe
X-ray film to detect probe/ RNA of interest
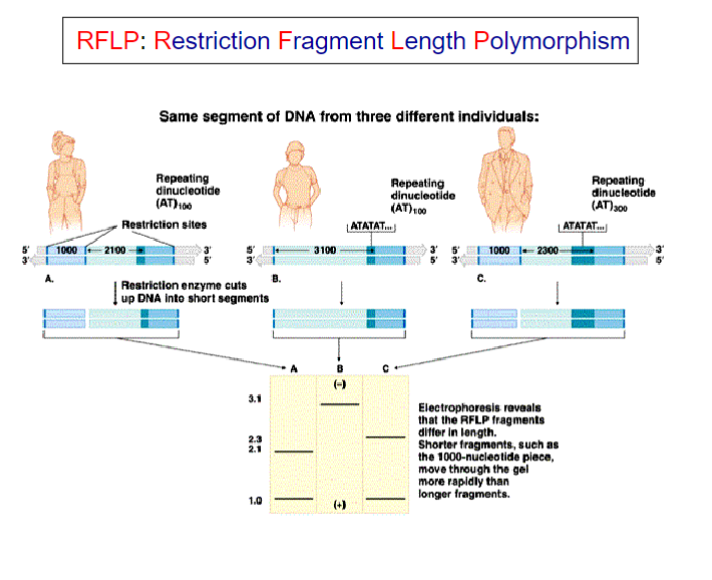
Restriction Fragment Length Polymorphisms [RFLP]
It refers to a difference between samples of homologous
DNA molecules that come from differing locations of restriction enzyme sites, and to a related laboratory technique by which these segments can be illustrated.
In RFLP analysis, the DNA sample is broken into pieces (digested) by restriction enzymes and the resulting restriction fragments are separated according to their lengths by gel electrophoresis.
Although now largely obsolete due to the rise of inexpensive DNA sequencing technologies, RFLP analysis was the first DNA profiling technique inexpensive enough to see widespread application.
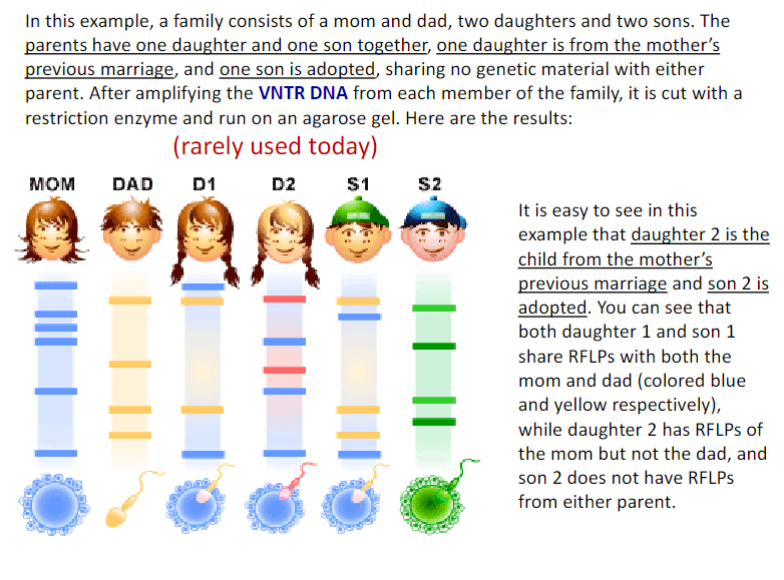 D1: child of these parents, D2: child of previous mother marriage, S1 child of these parents, S2: adopted son
D1: child of these parents, D2: child of previous mother marriage, S1 child of these parents, S2: adopted son
Variable Number Tandem Repeat (VNTR)
is a location in a genome where a short nucleotide sequence (10 -100 bp) is organized as a tandem repeat. These can be found on many chromosomes, and often show variations in length between individuals. Each variant acts as an inherited allele, allowing them to be
used for personal or parental identification.
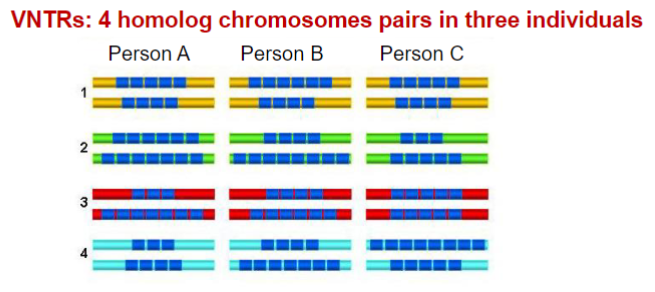
Minisatellites: variable number tandem repeats (VNTRs)
first polymorphisms used in DNA profiling
large amount of DNA was required
replaced by short tandem repeats (STRs)
Microsatellites: short tandem repeats (STRs)
STRs are currently the most commonly analyzed genetic polymorphism in forensic genetics
The general structure of VNTRs and STRs is the same

Single nucleotide polymorphisms (SNPs)
single base differences in the sequence of the DNA.
SNPs are formed when errors (mutations) occur as
the cell undergoes DNA replication.
However, SNPs are so abundant throughout the genome that it is theoretically possible to type hundreds of them
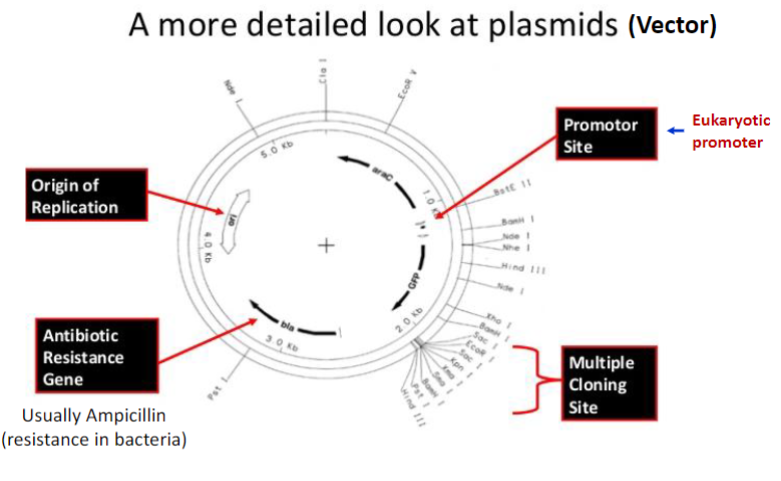
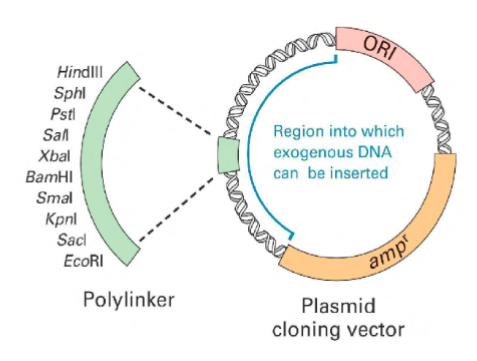
Vector:
A vector, in molecular biology, refers to a plasmid that is
engineered to make it a more useful tool for molecular biologists (all vectors are plasmids, but not all plasmids are vectors)
a multiple cloning site/region (MCS or MCR) – a short section of DNA containing several unique restriction enzyme recognition sequences
 Plasmid requirements for cloning:
Plasmid requirements for cloning:
antibiotic resistance gene
promoter region
multiple cloning sites for insertion of gene of interest
origin of replication
Selectable marker
genticin -neo cassette
puromycin- puro cassette
hygromycin B - hygro cassette
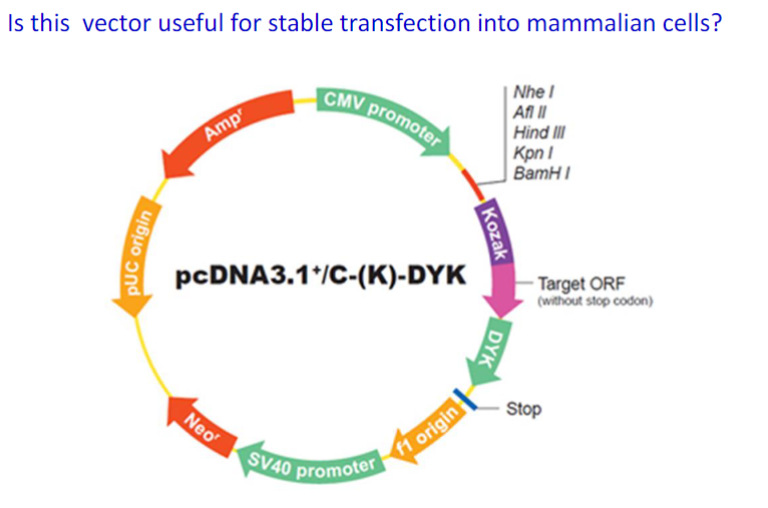 yes
yes
Subcloning
move a gene of interest from a parent vector to a destination vector
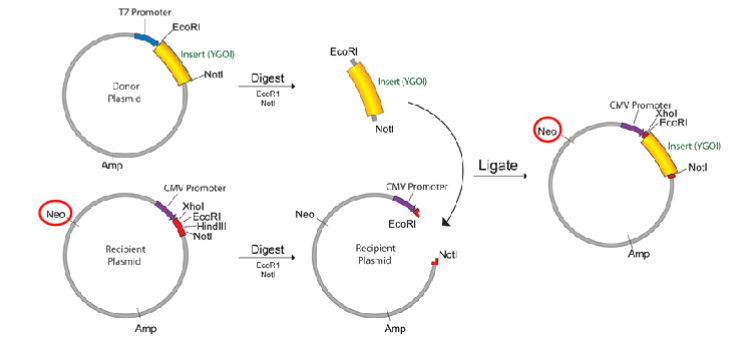
Recombinate DNA
To create recombinant DNAs scientist use cloning techniques.
That means creating a plasmid by transferring DNA fragments from one species to another and replicating the plasmid into bacteria.
The region where the new gene is inserted is called the "polylinker region" because a sequence of sites containing DNA recognized by different restriction enzymes has been artificially inserted
Because of the end complementarity, cutting two DNA s with the same enzyme makes possible annealing the two
fragments into one sequence, but without covalent linkage
Use of a DNA ligase could combine them
into a single sequence

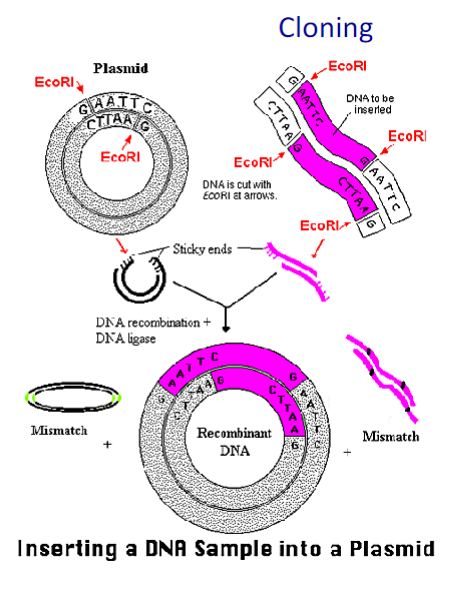
A typical DNA cloning experiment requires that the DNA to becloned (often called the “insert DNA”) and the vector (often a plasmid) both be cut with the same enzyme (or with two enzymes which produce compatible ends).
The insert DNA and the vector are then mixed, and DNA ligase is used to join the molecules.
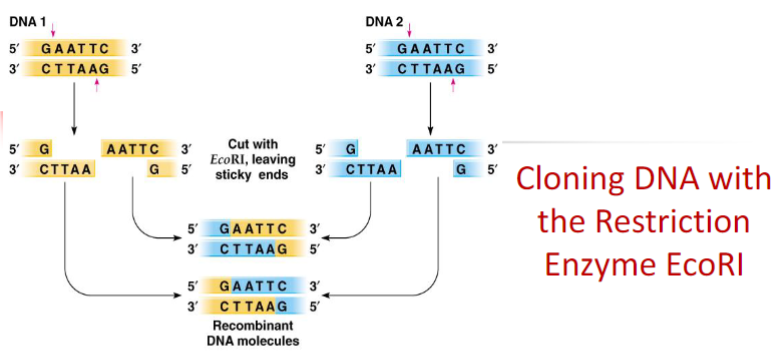
If two DNA fragments were cut with the same restriction enzyme (sticky end) it is possible to ligate those two different fragments. This is the base of cloning.
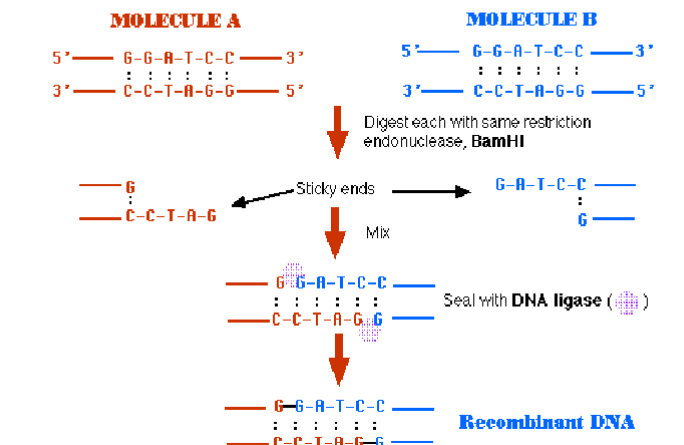
Blunt end ligation could be inserted in opposite direction.
It must be tested ligation in the right direction
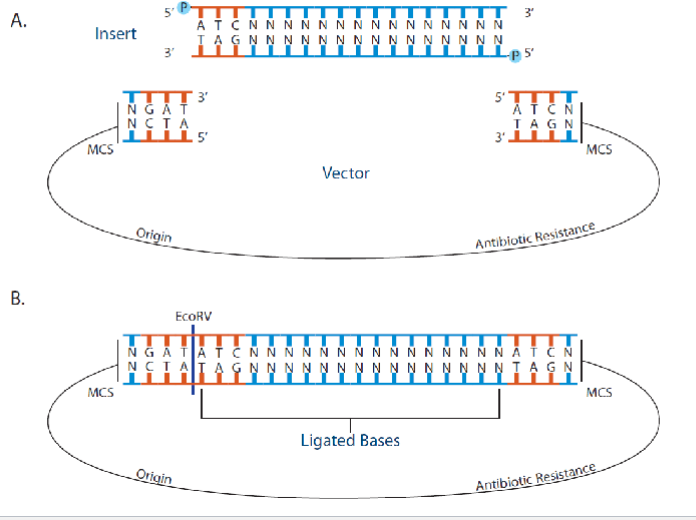
Cutting a plasmid with a single restriction enzyme (EcoRI in this case) cannot direct the correct orientation of the gene of interest to be inserted into the plasmid
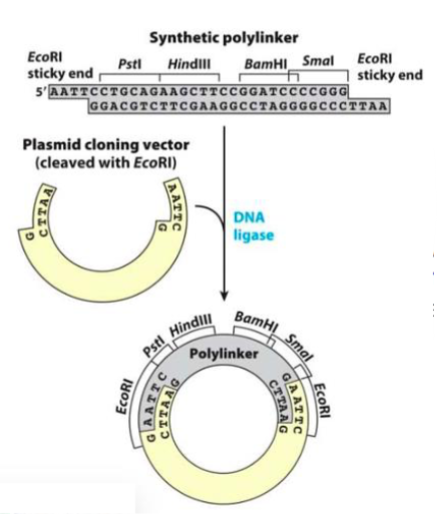
By using two different coherent restriction enzymes is
possible to insert new gene at the correct orientation
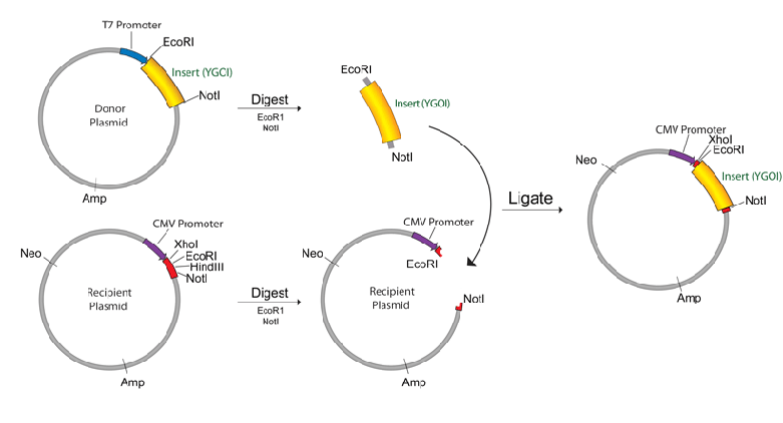
Instead plasmid it is possible to use viral vectors too
Lec soto 9 10/25 DNA Vectors Mutagenesis
Mammalian Expression Vector

Plasmids divide only in prokaryotic, not in eukaryotic cells
The number of copies of the plasmid depends on the ori sequence. Generally mutation in
ori results in loss of its regulation and increase in the number of copies
If a plasmid has too high of a copy number, they may excessively burden their host by occupying too much cellular machinery and using too much energy.
Low copy number plasmids have the advantage of reducing the metabolic burden imposed on the host cells. However, having a low copy number prevents them from relying on random diffusion for their distribution to daughter cells
Expression Vectors have variation in:
1. Selectable markers, antibiotic to select positive clones
2. Protein tags, small peptides incorporated into a translated protein
3. Promoters, constitutive or inducible
1. Selectable Markers
It is unlikely that all cells will take up the plasmid. Thus, many plasmids have selectable markers (neutralize antibiotic effect) to select only the cells that received the plasmid
Neomycin: protein synthesis inhibitor
Puromycin: protein synthesis inhibitor
Hygromycin: protein synthesis inhibitor
Seocin/Bleo : bind and cleave DNA
2. Epitope Tag or Fusion Protein
Fusing your protein to an epitope tag, such as Flag or HA, will allow you to easily identify your protein using an antibody against that epitope
can be used for western blots or immunoprecipitations without antibody
Another common scenario is fusing your protein to another protein, such as GFP, which allows you to visualize the cellular localization of your protein
FLAG-tag, or FLAG octapeptide, or FLAG epitope, is a polypeptide protein tag that can be added to a protein using recombinant DNA technology
Human influenza hemagglutinin (HA) tag has been extensively used as a general epitope tag in expression vectors
A myc tag is a polypeptide derived from the c-myc gene product
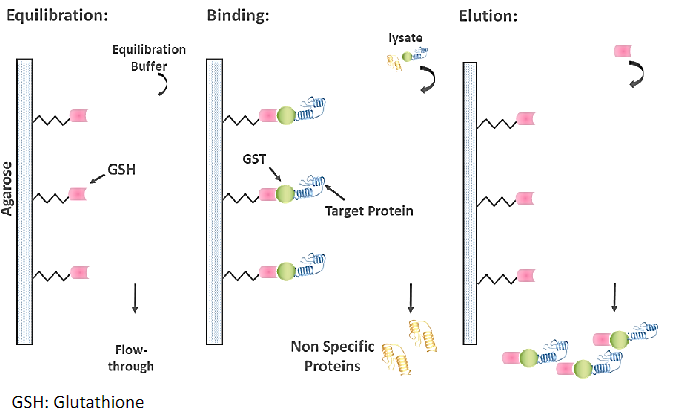
His-tag (Histidine) binds tightly to immobilized metal ions such as Nickel II (affinity chromatography)
used in protein purification like a bead and is eluted with Imidazole
GST, glutathione is a tripeptide (Glu-Cys-Gly). When reduced glutathione is immobilized through its sulfhydryl group to a solid support, such as cross- linked beaded agarose, it can be used to capture GST-tagged proteins
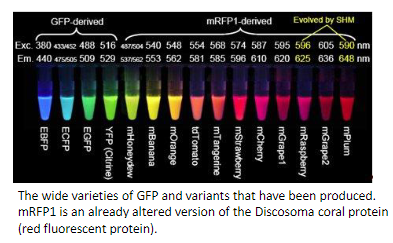
GFP, Green Fluorescent Protein that exhibits bright green fluorescence when exposed to light in the blue to ultraviolet range
3. Promoters
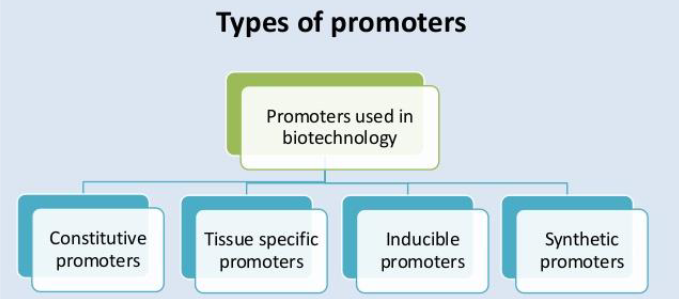
Constitutive promoters: These promoters direct expression in virtually all tissues and are largely, if not entirely, independent of environmental and developmental factors.
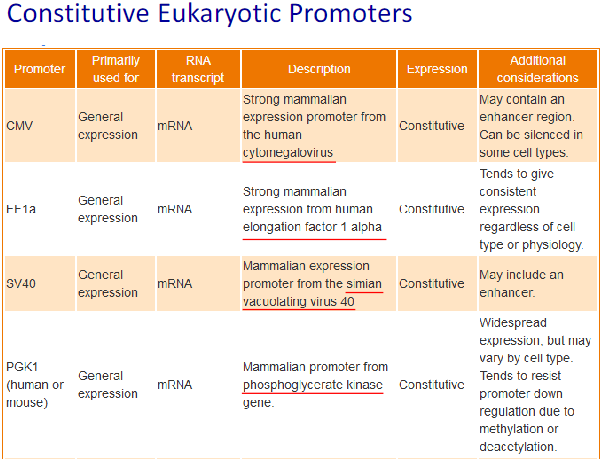
Tissue-specific promoters: These direct the expression of a gene in specific tissue(s) or at certain stages of development. Tissue or cell-specific promoters allow:
The expression of genes of interest into targeted organs or tissues
such as liver, brain, lung...
The discrimination or selection of a population of cells within a
tissue using fluorescent proteins (i.e. oligodendrocytes vs neurons)
To follow the differentiation process of living cells in a tissue
Inducible promoters: Their performance is not conditioned to endogenous factors but to external stimuli that can be artificially controlled. Promoters that respond to antibiotics, among other compounds, have been adapted to allow the induction of gene activity.
Tet-On system / Tet-Off system
Cumate system
Ecdysome system
Glucocorticoide system
Synthetic promoters: DNA sequences that do not exist in nature and which are designed to regulate the activity of genes
Tet-On system / Tet-Off system
Tet-On expression system
The Tet-On system is based on a reverse tetracycline-controlled transactivator, rtTA. Like
tTA, rtTA is a fusion protein comprised of the TetR repressor and the VP16 transactivation
domain; however, a four amino acid change in the tetR DNA binding moiety alters rtTA's
binding characteristics such that it can only recognize the tetO sequences in the TRE of the
target transgene in the presence of the Dox effector. Thus, in the Tet-On system,
transcription of the TRE-regulated target gene is stimulated by rtTA only in the presence
of Dox
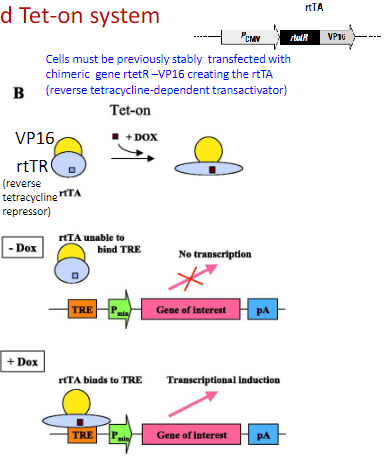 B: in contrast, addition of DOX to the Tet-on system (rtTA) results in transcriptional induction of the gene of interest. tTA, tetracycline-dependent transactivator; rtTA, reverse tetracycline-dependent transactivator; DOX, doxycycline (ligand); TRE, Tet-responsive element
B: in contrast, addition of DOX to the Tet-on system (rtTA) results in transcriptional induction of the gene of interest. tTA, tetracycline-dependent transactivator; rtTA, reverse tetracycline-dependent transactivator; DOX, doxycycline (ligand); TRE, Tet-responsive element
Components for a tetracycline on system are:
Plasmid containing a tetracycline-dependent promoter upstream of your gene of interest
rtTA or TetR expression plasmid
Stable cell lines can be made that continuously express a system component (e.g. tTA)
Tet-Off expression system
In the Tet-Off expression system, a tetracycline-controlled transactivator protein (tTA), which is composed of the Tet repressor DNA binding protein (TetR) from the Tc resistance operon of Escherichia coli transposon Tn10 fused to the strong transactivating domain of VP16 from Herpes simplex virus, regulates expression of a target gene that is under transcriptional control of a tetracycline-responsive promoter element (TRE). The TRE is made up of Tet operator (tetO) sequence concatemers fused to a minimal promoter, (commonly the minimal promoter sequence derived from the human cytomegalovirus (hCMV) immediate-early promoter). In the absence of Tc or Dox, tTA binds to the TRE and activates transcription of the target gene. In the presence of Tc or Dox, tTA cannot bind to the TRE, and expression from the target gene remains inactive
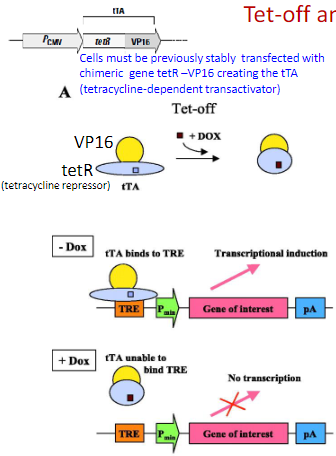 The Tet system can be used to conditionally activate gene expression. A: the Tet-off system (tTA) will activate expression in the absence of its ligand doxycycline (DOX, shown as brown box). Upon addition of DOX, transcription of the gene of interest is extinguished.
The Tet system can be used to conditionally activate gene expression. A: the Tet-off system (tTA) will activate expression in the absence of its ligand doxycycline (DOX, shown as brown box). Upon addition of DOX, transcription of the gene of interest is extinguished.
Basic components
Components for a tetracycline off system are:
Plasmid containing a tetracycline-dependent promoter upstream of your gene of interest
tTA expression plasmid
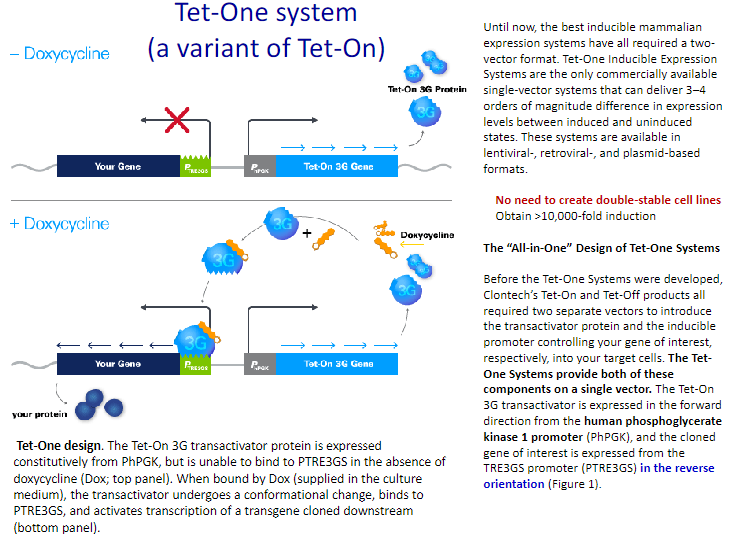
Cumate system:
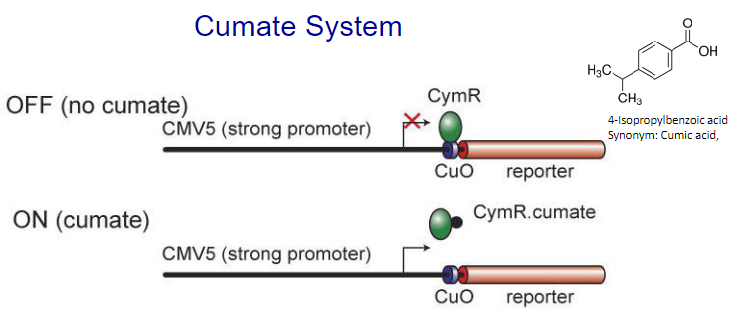 Schematic representation of the Cumate switch. Repressor configuration: The bacterial repressor, CymR, can bind to the operator sequence (CuO) placed downstream of CMV5, a strong viral promoter that is active in mammalian cells. Once bound, CymR blocks transcription from the CMV5 promoter. CymR bound to cumate is unable to bind to CuO. Transcription from CMV5 can proceed unhindered. Pseudomonas putida F1 utilizes p-cumate (p-isopropylbenzoate) as a growth substrate.
Schematic representation of the Cumate switch. Repressor configuration: The bacterial repressor, CymR, can bind to the operator sequence (CuO) placed downstream of CMV5, a strong viral promoter that is active in mammalian cells. Once bound, CymR blocks transcription from the CMV5 promoter. CymR bound to cumate is unable to bind to CuO. Transcription from CMV5 can proceed unhindered. Pseudomonas putida F1 utilizes p-cumate (p-isopropylbenzoate) as a growth substrate.
The Ecdysone system:
Ecdysone is a steroid hormone, which works in insects only.
One plasmid is expressing the Ecdysone receptor
The reporter plasmid contains the protein encoding sequence under the control of Ecdysone receptor.
This is a very tightly regulated system
Response to ecdysone is quantitative
Virtually no side effects
Ecdysone is metabolized similarly to other steroid hormones
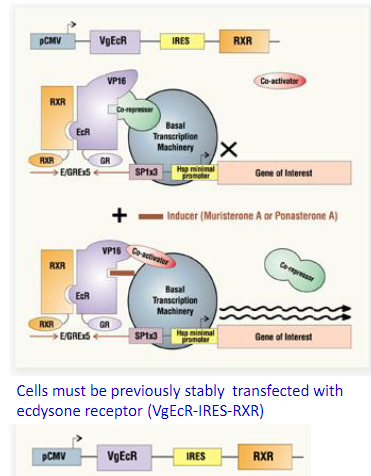 The Ecdysone-Inducible Expression System: The Nuclear receptor proteins RXR and VgEcR are coexpressed from the CMV promoter VgEcR: hybrid nuclear receptor comprised of the ecdysone receptor (EcR) ligand-binding and dimerization domains, the VP16 transcriptional activation domain and the glucocorticoid receptor (GR) DNA binding domain. The heterodimeric ecdysone receptor remains bound to five copies of the E/GRE recognition element located upstream of a minimal promoter in the inducible expression cassette. The inducible promoter remains transcriptionally silent until induction with the ecdysone analogs muristerone A or ponasterone A. Interaction between the inducer and the EcR ligand-binding domain results in the recruitment of coactivator(s) and, thus, transcriptional activation that can reach over three orders of magnitude.
The Ecdysone-Inducible Expression System: The Nuclear receptor proteins RXR and VgEcR are coexpressed from the CMV promoter VgEcR: hybrid nuclear receptor comprised of the ecdysone receptor (EcR) ligand-binding and dimerization domains, the VP16 transcriptional activation domain and the glucocorticoid receptor (GR) DNA binding domain. The heterodimeric ecdysone receptor remains bound to five copies of the E/GRE recognition element located upstream of a minimal promoter in the inducible expression cassette. The inducible promoter remains transcriptionally silent until induction with the ecdysone analogs muristerone A or ponasterone A. Interaction between the inducer and the EcR ligand-binding domain results in the recruitment of coactivator(s) and, thus, transcriptional activation that can reach over three orders of magnitude.
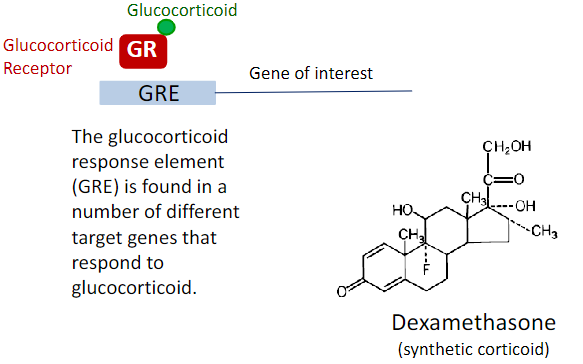
Glucocorticoid Inducible Promoters:
The biological actions of glucocorticoids are mediated by binding to specific glucocorticoid receptors (GRs)
Synthetic Promoters:
Obtain expression data (e.g. microarray, RNA-seq) under representative culturing conditions. Parse, process, and analyze expression data with a statistical mode (e.g. mixture model)
Identify genes from different expression groups and annotate putative transcription facto binding sites (TFBSs) upstream of the transcribed region for each gene
Build synthetic promoters using TFBS frequencies analyzed from annotation and expression data between groups
Test de novo, synthetic promoter designs with key TFBSs
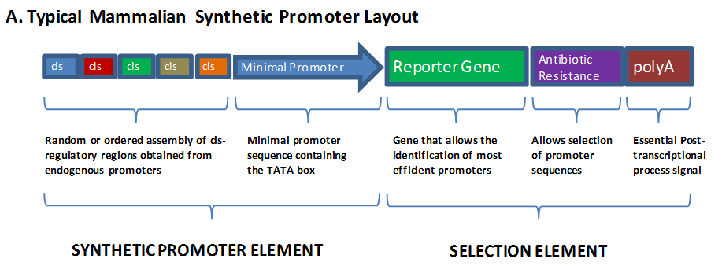 In recent years some efforts have been made to construct synthetic promoters for tissue specific transcription based on the linking of short oligonucleotide promoter and enhancer elements in a random or ordered fashion.
In recent years some efforts have been made to construct synthetic promoters for tissue specific transcription based on the linking of short oligonucleotide promoter and enhancer elements in a random or ordered fashion.
Mutagenesis:
DNA mutations deliberately engineered to produce mutant genes, proteins, strains of bacteria, and other genetically modified organisms
Focus for class is on PCR techniques that are produced by mutations and deletions
Site-directed Mutagenesis:
Site-directed mutagenesis creates a mutation at a defined site and requires a known template sequence
For the alteration, SNPs single nucleotide polymorphisms may be used to insert or delete a sequence element (ligand binding site or restriction site
Site-directed mutagenesis is typically performed using PCR
A primer that contains the complementary bases for the desired sequence (mutation) is used in a PCR
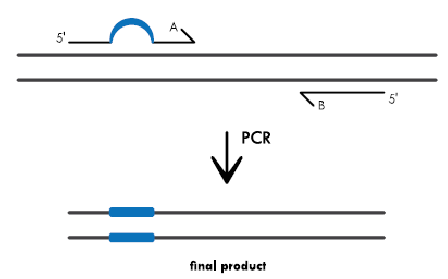 PCR for Base Substitutions: Primers containing the base changes of interest as a non-complementary break in the primer sequence (indicated by the blue bubble in primer A) are used in a PCR reaction. As the primers are extended, the resulting amplification product incorporates the mutation, replacing the original sequence (shown as a blue bar in the PCR product)
PCR for Base Substitutions: Primers containing the base changes of interest as a non-complementary break in the primer sequence (indicated by the blue bubble in primer A) are used in a PCR reaction. As the primers are extended, the resulting amplification product incorporates the mutation, replacing the original sequence (shown as a blue bar in the PCR product)
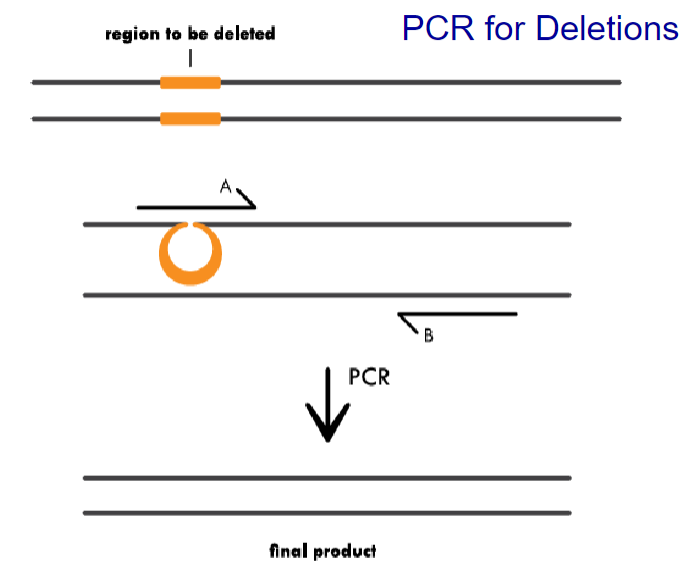 PCR for Deletions: Primer A contains complementary sequence to the regions flanking the area to be deleted. During PCR, primer binding will cause a region of the template to loop out, and amplify only the complementary region. The final product is shorter because it is missing the deleted sequence
PCR for Deletions: Primer A contains complementary sequence to the regions flanking the area to be deleted. During PCR, primer binding will cause a region of the template to loop out, and amplify only the complementary region. The final product is shorter because it is missing the deleted sequence
Variation of Mutagenesis: Inverse PCR for a Deletion
Inverse PCR for a Deletion. This method uses primers that hybridize to regions on either side of the area to be deleted. In this case, the primers contain 5’ phosphorylated ends to allow the two ends to be ligated together following amplification. PCR with a high fidelity DNA polymerase that leaves blunt ends creates a linearized fragment that is missing the deleted region. This fragment is then recircularized by intramolecular ligation and the resulting plasmid is transformed
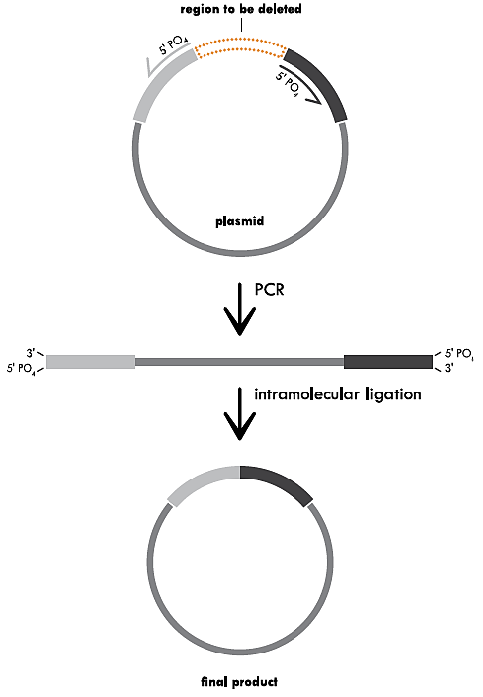 Inverse PCR for a Substitution
Inverse PCR for a Substitution
Inverse PCR for a Substitution. One of the two primers contains the mutation (substitution sequence) of interest (indicated by the blue bubble). In this case, both primers contain 5’ phosphorylated ends to allow the two ends to be ligated together following amplification. PCR is used to amplify the entire circular plasmid to create a linear template that contains the substituted sequence. This fragment is then recircularized by intramolecular ligation and the resulting plasmid is transformed
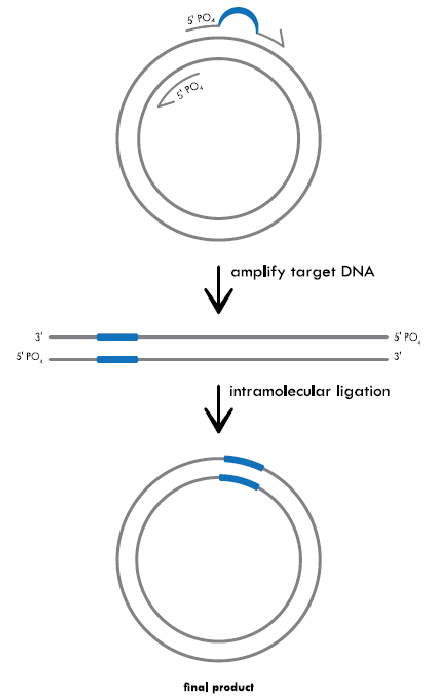
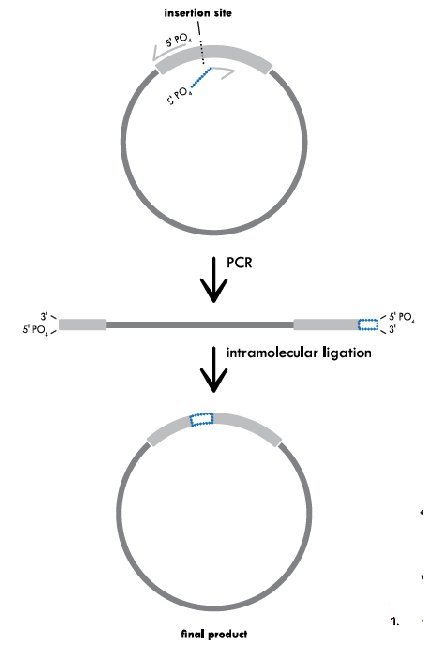
Inverse PCR for an Insertion:
The primers are lined up back-to-back on either side of the area where the new sequence will be inserted (indicated by the black, dotted line). One of the primers contains the additional sequence that will be inserted (indicated by the blue line). Both primers contain 5’ phosphorylated ends to facilitate ligation following amplification. PCR creates a linearized fragment containing the new sequence. The plasmid is then recircularized by intramolecular ligation and transformed
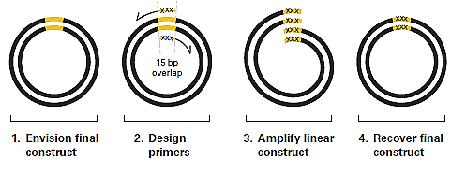
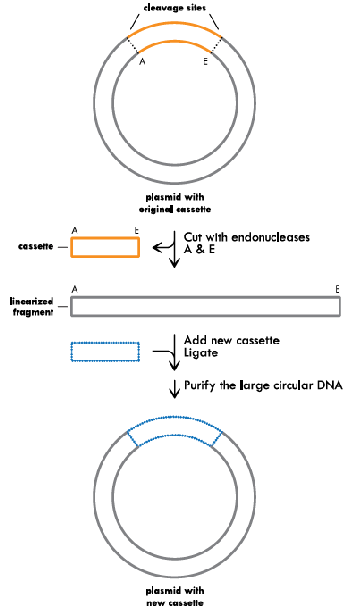
Cassette Mutagenesis:
The original plasmid is cleaved with restriction enzymes A and E on either side of the cassette to be removed (indicated in orange). The restriction digest creates a linearized plasmid fragment and a cassette. The new cassette (indicated in blue) containing the desired changes is then ligated into the linearized plasmid
Summary:
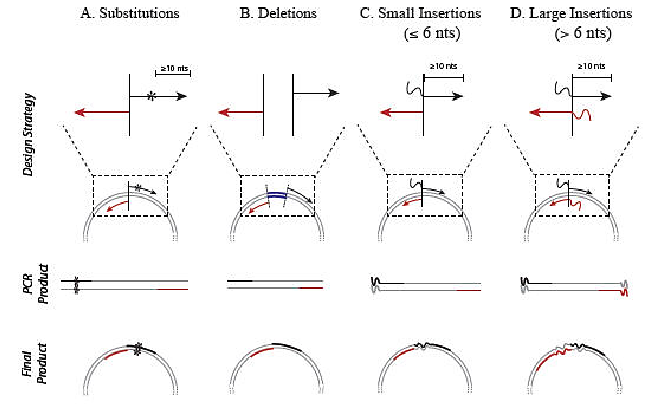 Substitutions, deletions and insertions are incorporated into plasmid DNA through the use of specifically designed forward (black) and reverse (red) primers. Unlike kits that rely on linear amplification, primers designed for the Q5 Site-Directed Mutagenesis Kit should not overlap to ensure that the benefits of exponential amplification are realized. A) Substitutions are created by incorporating the desired nucleotide change(s) (denoted by *) in the center of the forward primer, including at least 10 complementary nucleotides on the 3 ́side of the mutation(s). The reverse primer is designed so that the 5 ́ ends of the two primers anneal back-to- back. B) Deletions are engineered by designing standard, non-mutagenic forward and reverse primers that flank the region to be deleted. C) Insertions less than or equal to 6 nucleotides are incorporated into the 5 ́ end of the forward primer while the reverse primer anneals back-to-back with the 5 ́ end of the complementary region of the forward primer. D) Larger insertions can be created by incorporating half of the desired insertion into the 5 ́ ends of both primers. The maximum size of the insertion is largely dictated by oligonucleotide synthesis limitations
Substitutions, deletions and insertions are incorporated into plasmid DNA through the use of specifically designed forward (black) and reverse (red) primers. Unlike kits that rely on linear amplification, primers designed for the Q5 Site-Directed Mutagenesis Kit should not overlap to ensure that the benefits of exponential amplification are realized. A) Substitutions are created by incorporating the desired nucleotide change(s) (denoted by *) in the center of the forward primer, including at least 10 complementary nucleotides on the 3 ́side of the mutation(s). The reverse primer is designed so that the 5 ́ ends of the two primers anneal back-to- back. B) Deletions are engineered by designing standard, non-mutagenic forward and reverse primers that flank the region to be deleted. C) Insertions less than or equal to 6 nucleotides are incorporated into the 5 ́ end of the forward primer while the reverse primer anneals back-to-back with the 5 ́ end of the complementary region of the forward primer. D) Larger insertions can be created by incorporating half of the desired insertion into the 5 ́ ends of both primers. The maximum size of the insertion is largely dictated by oligonucleotide synthesis limitations
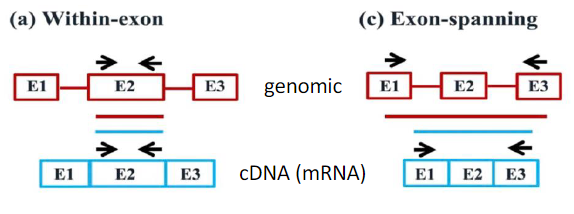
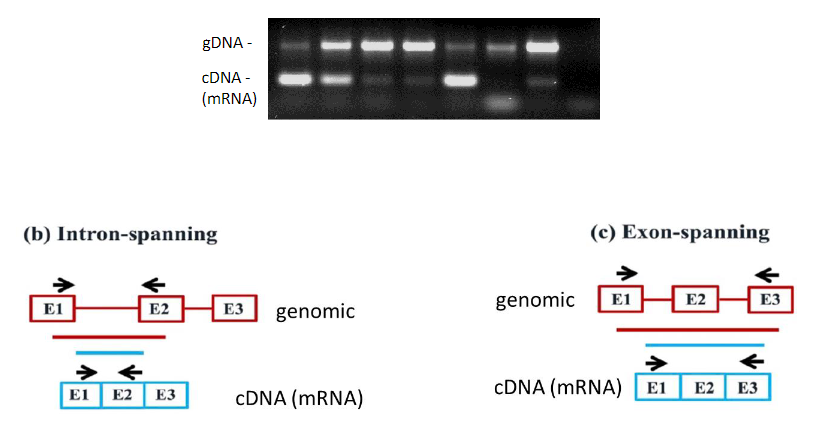
The impact of RT-qPCR primer design strategy on potential amplification products from cDNA and gDNA:
Schematic representation of gDNA (in red) showing exons (boxes) and introns (lines), and cDNA (in blue) showing exons (boxes). The primer locations over cDNA and gDNA (black arrows) and the resulting PCR amplicon from gDNA (red bar) and cDNA (blue bar) are illustrated. (a) Within-exon primers: This primer design will generate undistinguishable amplification products from both cDNA and contaminating gDNA templates. (b) Intron-spanning primers: In addition to an amplicon of the expected size from cDNA, this primer design can also generate a longer amplicon from gDNA if the size of the spanned intron is sufficiently short (see Fig. 5). (c) Exon spanning primers: This primer design strategy that can be used to detect the splicing of cassette exons in cDNA (Padhi and Pelletier, 2012) may still generate longer amplicons from gDNA if the sizes of the spanned exon and introns are short. (d) Exon-exon junction primers: This primer design will amplify sequences from cDNA but not from gDNA