CFTR and Cystic Fibrosis
Overview of Cystic Fibrosis (CF):
Definition and Genetics:
Cystic Fibrosis (CF) is a monogenic autosomal recessive disorder caused by mutations in a single gene: CFTR (Cystic Fibrosis Transmembrane Conductance Regulator).
Autosomal recessive inheritance:
Individuals must inherit two defective CFTR alleles (one from each parent) to develop CF.
Carriers with one functional and one mutant allele are typically asymptomatic.
Gene location: CFTR is located on chromosome 7q31.2.
Prevalence:
Common among individuals of Northern European ancestry.
In the UK, approximately 1 in 2,400 live births are affected.
Rare in populations such as those of East Asian descent (e.g. Japan, which is 1 in 350,000).
Organs Affected and Clinical Manifestations:
Organs Involved:
Respiratory system – chronic lung infections and progressive respiratory failure.
Pancreas – pancreatic insufficiency leading to malabsorption and steatorrhoea.
Gastrointestinal tract – intestinal obstruction (e.g. meconium ileus in neonates).
Reproductive system – male infertility due to congenital bilateral absence of the vas deferens (CBAVD).
Sweat glands – elevated sweat chloride concentration.
Liver – biliary cirrhosis in advanced cases.
Pathophysiological Basis:
Defective CFTR-mediated ion transport causes abnormal chloride (Cl⁻) and bicarbonate (HCO₃⁻) secretion across epithelial surfaces.
Which leads to:
Reduced airway surface liquid (ASL) due to increased sodium (Na⁺) reabsorption via ENaC (epithelial sodium channel).
Dehydrated, viscous mucus, impairing mucociliary clearance.
Chronic bacterial infection and inflammation, especially in the lungs.
Classification:
Classic CF:
High sweat chloride (>60 mmol/L).
Multisystem involvement (pancreas, lungs, etc.).
More severe clinical course and reduced life expectancy.
Non-classic CF:
Borderline/normal sweat chloride.
Often single organ involvement.
Milder symptoms, sometimes diagnosed later in life.
CFTR Structure and Function:
CFTR Overview:
The Cystic Fibrosis Transmembrane Conductance Regulator.
CFTR is a cAMP-regulated anion channel belonging to the ATP-Binding Cassette (ABC) transporter superfamily.
Expressed on the apical membrane of epithelial cells in organs such as the lungs, pancreas, intestines, and reproductive ducts.
Structural Features:
Composed of 1,480 amino acids forming a large membrane protein.
Domain architecture:
Two transmembrane domains (TMD1, TMD2) – each contains six α-helical membrane-spanning segments forming the ion pore.
Two nucleotide-binding domains (NBD1, NBD2) – bind and hydrolyse ATP to regulate gating.
Regulatory (R) domain – unique to CFTR; controls channel opening via phosphorylation by protein kinase A (PKA).
Long N- and C-terminal extensions – contribute to protein stability and regulatory interactions.
Glycosylation occurs on the fourth extracellular loop, critical for correct folding and membrane localisation.
Mechanism of Channel Regulation:
Phosphorylation-dependent gating:
PKA phosphorylates multiple serine residues in the R domain (9 consensus sites).
This reduces the R domain’s affinity for NBDs, allowing ATP binding and channel opening.
ATP binding and hydrolysis:
Once the R domain is phosphorylated, ATP molecules can bind to both NBD1 and NBD2. The binding of ATP at both sites triggers a conformational change that brings the two NBDs together, forming a "head-to-tail" dimer.
This NBD dimerisation is coupled to a conformational change in the TMDs, which leads to the opening of the ion channel pore, allowing Cl⁻ and HCO₃⁻ ions to flow down their electrochemical gradients.
ATP is then hydrolysed, primarily at NBD2, but both NBDs contribute to the overall cycle. The energy from ATP hydrolysis is used to break the NBD dimer.
This causes the NBDs to separate, and the channel pore closes.
Ion conductance:
Facilitates secretion of Cl⁻ and HCO₃⁻ ions.
This ion movement creates an osmotic gradient, which hydrates the airway surface liquid, thinning mucus, and enables effective mucociliary clearance.
Inhibits ENaC, thereby reducing Na⁺ absorption and decreases water reabsorption.
Consequences of CFTR Dysfunction:
Loss of CFTR Function:
Impaired Cl⁻ and HCO₃⁻ secretion.
Increased Na⁺ absorption (due to disinhibition of ENaC).
Leads to dehydrated mucus and impaired mucociliary clearance.
Mucociliary Clearance and Lung Pathophysiology:
In normal lungs:
Cilia beat in a well-hydrated periciliary layer (PCL), propelling mucus out at ~60 μm/s.
In CF lungs:
Hyperabsorption of airway surface fluid causes the mucus to collapse onto cilia.
Cilia cannot beat effectively → impaired clearance of pathogens.
Creates a hypoxic, acidic environment ideal for bacterial colonisation (e.g. Pseudomonas aeruginosa).
Inflammation and Tissue Damage:
Persistent infection triggers chronic neutrophilic inflammation.
Excessive release of proteases (e.g. elastase) damages epithelial cells.
Cytokine imbalance (↑ IL-8, TNF-α) perpetuates inflammation.
Results in bronchiectasis, fibrosis, and progressive decline in lung function.
Molecular Basis of CFTR Mutations:
Diversity of Mutations:
Over 1,950 CFTR mutations identified.
Categorised by their effect on CFTR synthesis, processing, and function.
Including deletions, missense, frameshift and nonsense mutations.
Notable Mutations:
ΔF508 (F508del):
Most common (70% homozygous; 90% at least one allele).
Located in NBD1.
Causes both Class II (misfolding) and Class III (gating) defects, and sometimes Class VI (stability).
Infection and Inflammation in CF Lungs:
Primary Causes:
Loss of CFTR function leads to:
Dehydrated mucus leads to impaired mucociliary clearance.
Acidic airway surface liquid (ASL) due to loss of HCO₃⁻ secretion.
The acidic environment impairs innate immune factors (e.g. lysozyme, defensins).
Resulting Pathology:
Accumulation of viscous mucus supports bacterial growth.
Chronic infection leads to sustained neutrophil recruitment.
Excess DNA and actin from lysed neutrophils further increase mucus viscosity.
Progressive airway remodelling which leads to a loss of pulmonary function and respiratory failure (leading cause of mortality).
Current Therapies (Symptomatic Management):
Traditional Treatments:
Antibiotics – control chronic bacterial infection (e.g. P. aeruginosa).
Anti-inflammatory agents – reduce tissue damage (e.g. corticosteroids, azithromycin).
Mucolytics – decrease mucus viscosity:
Dornase alfa (DNase I): breaks down extracellular DNA from neutrophils.
Physiotherapy – assists in mucus clearance.
Nutritional supplements – pancreatic enzyme replacement and high-calorie diet.
Impact:
Increased median life expectancy from ~5 years (1970s) to 35–40 years today.
Improved quality of life despite lack of curative treatment.
Gene Therapy Approaches:
Concept:
Aim: introduce a functional CFTR gene to restore ion channel activity.
Types of Gene Therapy:
Germline therapy – modifies germ cells (heritable); unethical and illegal in humans.
Somatic therapy – modifies somatic cells; non-heritable and currently permitted.
Delivery Strategies:
In vivo – direct delivery into bloodstream; systemic distribution but high immunogenicity.
In situ – targeted delivery to the affected organ (e.g. nasal spray for lungs).
Ex vivo – cells modified outside the body and reintroduced.
Gene Delivery Approaches:
Gene addition – insertion of functional CFTR transgene (risk of insertional mutagenesis).
Gene correction (genome editing) – precise repair of mutation using CRISPR/Cas systems.
Gene knockdown – for diseases of overexpression (less relevant for CF).
Vectors:
Viral vectors: Adenovirus, AAV, Lentivirus (high efficiency but immune risk).
Non-viral vectors: Liposomes, nanoparticles (lower efficiency, safer).
Despite >20 trials, no significant therapeutic success yet achieved due to:
Airway barriers (mucus layer, inflammation).
Limited access to lower lungs.
Unclear optimal target cell type.
Risk of immune response and limited expression duration.
Small Molecule CFTR Modulators
Small, orally active molecules that target specific CFTR defects.
Aim to restore CFTR function rather than treat symptoms.
Types of Modulators:
Production Correctors (Class I)
Promote read-through of premature stop codons.
Example: Ataluren (PTC124).
Correctors (Class II)
Improve CFTR folding and trafficking.
Example: Lumacaftor (VX-809).
Potentiators (Class III)
Increase open probability (Po) of the channel.
Example: Ivacaftor (VX-770).
Combination Therapies
Combine corrector + potentiator for compound mutations like ΔF508.
Example: Lumacaftor/Ivacaftor (Orkambi).
Key Case: Ivacaftor (VX-770):
Developed via high-throughput screening of >230,000 compounds.
Restores CFTR channel activity in G551D mutants (~50% of wild-type conductance).
Clinical outcomes:
Improved FEV₁ (lung function).
Decreased sweat chloride levels.
Enhanced quality of life.
Effective in ~5% of CF patients (those with gating mutations).
Key Case: Lumacaftor (VX-809):
Improves folding and trafficking of ΔF508 CFTR.
Alone: modest effect (~15% of wild-type activity).
Combined with Ivacaftor → synergistic increase (~25% activity).
Demonstrates multi-drug correction for complex alleles.
just written by ai
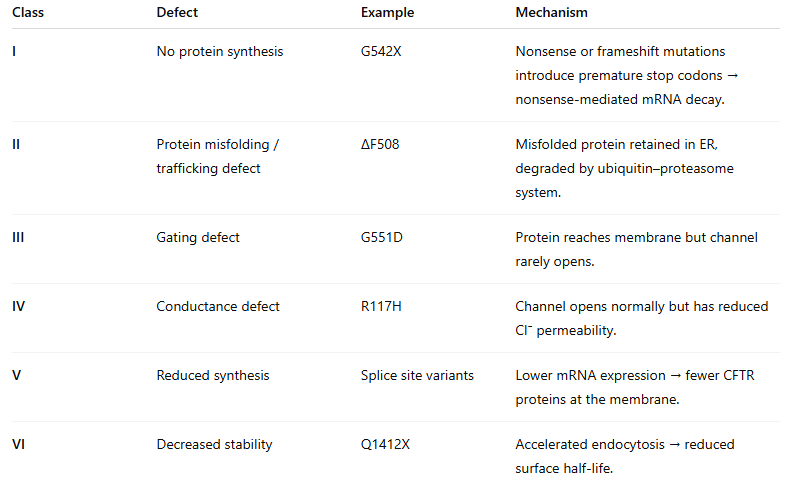
Class I Mutations: No CFTR Protein Production
Result: Complete absence of CFTR protein, meaning no protein is made at all.
Mechanism: These mutations typically introduce premature 'stop' signals (stop codons) in the genetic code.
This leads to nonsense-mediated decay (NMD) of the messenger RNA (mRNA), which is a quality control mechanism in the cell that degrades faulty mRNA, thus preventing any protein production.
Examples:
Nonsense Mutations (e.g., G542X): A single point mutation changes a normal codon into a premature stop codon, resulting in a severely shortened and non-functional CFTR protein. The mRNA is often 'chucked out' by NMD.
Frameshift Mutations (insertions/deletions): Small insertions or deletions of DNA 'letters' (nucleotides) that are not in multiples of three. This completely 'messes up' the reading frame downstream of the mutation, leading to completely different amino acids and usually an early stop codon. This faulty mRNA is also a target for NMD.
Splice Site Mutations: Alterations in the DNA sequence that affect how the pre-mRNA is 'edited' (spliced). This can lead to 'missing bits' (exon skipping) or 'extra bits' (intron retention), both of which can cause frameshifts and premature stop codons.
Class II Mutations: Defective Protein Processing and Trafficking
Result: The CFTR protein is made, but it's 'faulty' – it misfolds and gets stuck in the endoplasmic reticulum (ER).
Mechanism: Due to its incorrect folding, the protein fails the 'quality control' checks in the ER. It's then tagged for degradation and 'chucked out' by the cell's waste disposal system (the proteasome), so it never makes it to the cell surface.
Example:
F508del: This is a deletion of three nucleotides that removes a phenylalanine amino acid at position 508. This is the most common CF mutation and is a prime example of a misfolding error.
Class III Mutations: Defective Channel Gating
Result: The CFTR protein makes it to the cell surface, but the 'gate' of the channel won't open, or only opens very rarely.
Mechanism: Even when the necessary energy molecule (ATP) is present, the channel struggles to open effectively, severely limiting the flow of ions.
Example:
G551D: In this mutation, glycine at position 551 is replaced by aspartic acid. The protein is in the right place, but the channel simply doesn't 'flick open' properly.
Class IV Mutations: Defective Channel Conductance
Result: The CFTR protein reaches the cell surface and its 'gate' can open, but it's 'sluggish' – it can't conduct chloride ions through the channel efficiently.
Mechanism: The channel is active, but the actual flow of ions through it is significantly reduced.
Example:
R117H: Arginine at position 117 is replaced by histidine. The channel opens, but the 'stream' of chloride ions passing through is much weaker.
Class V Mutations: Reduced Protein Production
Result: Not enough functional CFTR protein is produced.
Mechanism: This can happen for various reasons, such as partially faulty splice sites, which means some normal mRNA is made alongside abnormal mRNA, or issues in the 'start' regions (promoters) of the gene that affect how much protein is transcribed.
Some functional CFTR is present, but it's well below the normal amount needed.
Example:
3849+10kb C->T: This 'deep intronic' splice mutation creates an alternative, 'dodgy' splice site. This results in a mix of correctly spliced CFTR mRNA (about 10%) and incorrectly spliced mRNA, leading to reduced overall functional protein.
Class VI Mutations: Accelerated Protein Degradation at the Cell Surface
Result: The CFTR protein reaches the cell surface and works for a bit, but it's 'short-lived' and quickly degraded.
Mechanism: These mutations often affect the 'tail end' (C-terminus) of the protein. This part of the protein is important for its stability and how long it stays on the cell membrane, leading to a rapid loss of functional channels.