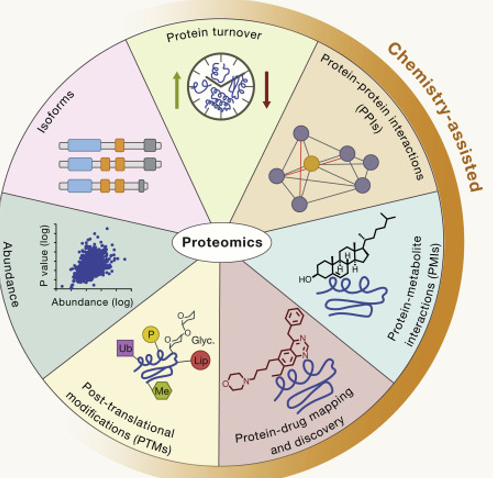
Proteomics
Proteomics
It is the large-scale study of proteins, particularly their structures, functions and interactions
Proteins are vital parts of living organism as they are the main components of the physiological metabolic pathways of cells
The proteome encompasses the proteins of an entire system
Cell, Tissue or Organism
Including modification
The proteome is dynamic
It varies with
Time
Responses to the environment
During development and differentiation
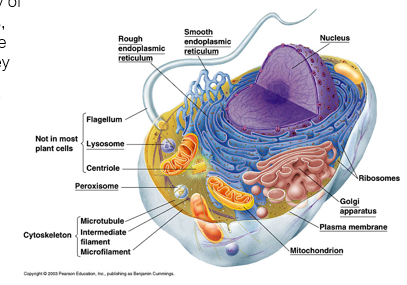
Complexity of the proteome
Genome : 20 - 25000 genes
Population variations in alleles
Mrna
Estimated that 40-60% can be alternatively spliced
RNA editing
Mainly Cytidine> Uracil
Adenine>Inosine
RNA splicing estimates vary
RNA editing: C>U - prime example is Apolipoprotein B: intestine produces a transcript with a CAA edited to UAA (stop codon), yielding a shorter form than the liver produces.
PTMs
Cleavage
Phosphorylation
Acetylation
Glycosylation
Lipidation
(30+ PTMS reported)
Protein-protein/protein-nucleic acid interactions
Receptors
Metabolic pathways
Signalling complexes
Transcription activation
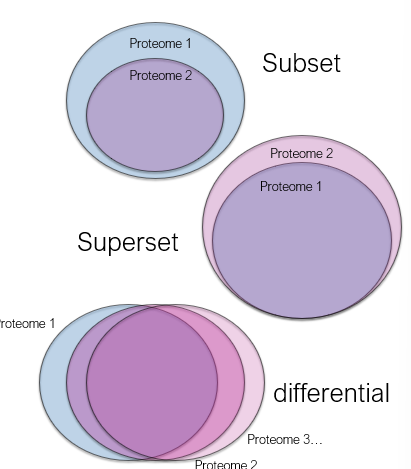
Comparing proteomes
Proteome size in that
Cell
Tissue
Organism
Comparison with proteome
Under different conditions
After differentiation
Tumour stage
Drug treatment
Changes in protein
Abundance
Activity
Modification
Interactions
Locations
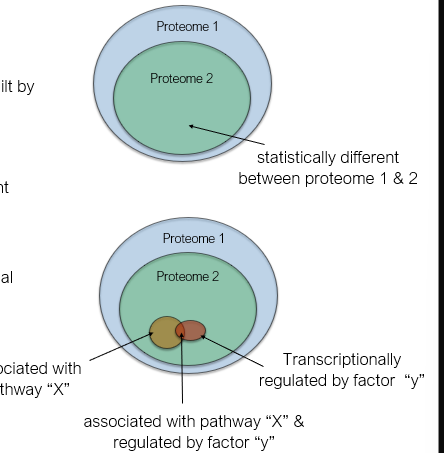
Comparing proteomes
Data mining to find relevant links (guilt by association)
All proteins which show alteration - statistical rigour
Proteins associated with a relevant
Protein complex
Signal transduction pathway
Product of shared transcriptional regulation
Methods for exploring the proteome
Classical Protein Biochemistry
Purification
Activity
Structure determination
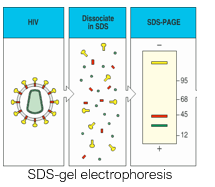
SDS-Polyacrylamide gel electrophoresis
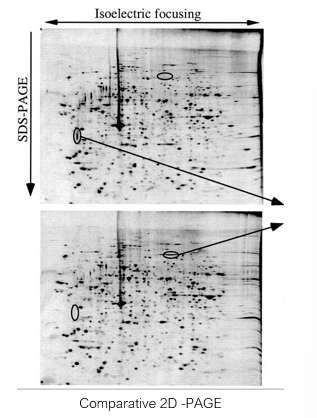
Two dimensional gel electrophoresis(size and charge)
Differential 2D gel electrophoresis
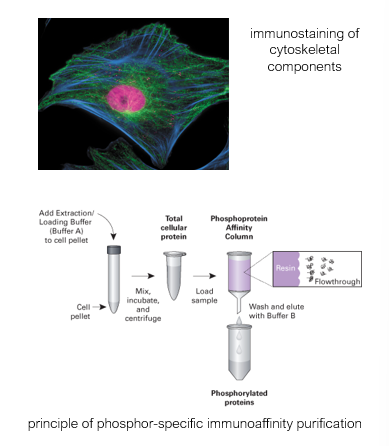
Immunoaffinity techniques
Localisation/Co-localisation
Purification
Single proteins
Functional groups
Complexes
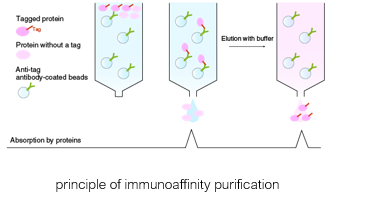
Protein-protein interactions
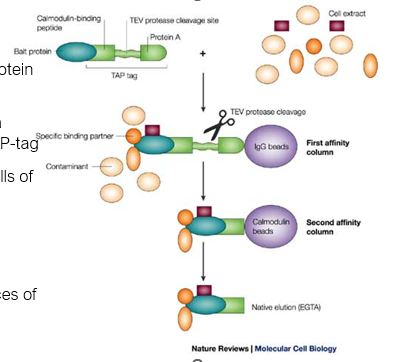
Tandem-affinity tags for protein complex analysis
Tag the DNA sequence of a protein of interest with a TAP-tag
Express fusion protein in cells of interest
Purify
Identify associated proteins
Repeat using DNA sequences of purified proteins
Build associated network
Tap-Tags are one of many ways proteins can be tagged with other molecules to help isolate or report on interaction partners.
Other options include yeast two-hybrid and associated techniques
Tap tagging can be done on a large scale
How can we identify proteins
Historically by extensive purification and characterisation
Amino acid sequencing
Now - Mass spectrometry methods
Direct identification/sequencing of purified protein fragments
From mixtures of proteins
Require existing knowledge of protein sequences
Has benefitted from
Genome initiatives
Bioinformatics of protein coding regions
Sequence databases
Protein sequence databases
Sites
Expasy - Geneva
European Bioinformatics Institute - EBI; Cambridge
NCBI - Bethesda, USA
Content
Directed sequenced protein data
Protein sequence derived from Genomic/mRNA sequences
Variants
Modifications
Key points
Comprehensive and curated
Non-redundant
Taxonomic
Internet accessible/searchable
Protein Mass Spectrometry - has two key methods
Electrospray Ionisation (ESI) | Matrix-assisted, laser desorption/Ionisation(MALDI) |
John B. Fenn | Kochi Tanaka |
2002 Nobel prize in Chemistry | 2002 Nobel prize in Chemistry |
Analysis of ionised peptide/proteins | Analysis of ionised peptide/proteins by mass spectrometry |
Applying a high voltage to an aerosol nozzle through which protein solution is passed | Protein/Peptides dried with an acid matrix compound |
Solvent evaporates in vacuum of mass spectrometer, leaving charge on the biomolecules | UV laser light causes ablation(desorption) of matrix and peptides |
Very good for large molecules | Matrix helps transfer protons(+-H) to the peptides |
Gives high quality information when combined with Tandem mass spectrometry(ESI-MS-MS) | Ionised peptides can be examined for mass to charge ratio(m/z) in a time of flight spectrometer(TOF) |
Can de novo sequence peptides |
|
Predominantly multiple ionisations | Predominantly single ionisation |
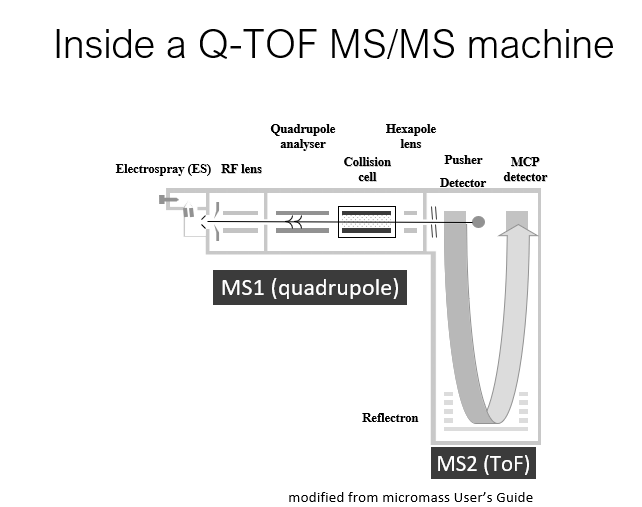
Mass Spectrometry Terms
Mass analysers
Time of flight(TOF) mass analyser
Accelerates charged ions in a vacuum tube, measuring the flight time
Quadrupole mass filters
Uses radio frequencies and DC voltages to filter ions based on their mass to charge(m/z) ratio
Collision cell
A device which enables collision activated dissociation of peptides into smaller fragments
Usually by collision with an inert gas
Ion optics
Ion lenses
Focus ions into an appropriate beam
Reflection
Mirror for ions, used to extend flight time
Ion detector
Detects ions striking it, amplifies signal
Ion Traps
Similar to quadrupole filters, but capable of trapping and accumulating a chosen ion
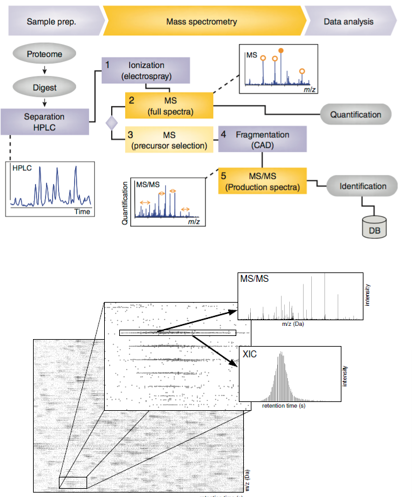
Automation : LC-MS/MS
Whole proteomes are complex mixtures
Increasingly so when Trypsin is fragmented
Too many ions
Pre-sort ions using Liquid chromatography(LC)
Ion exchange column
Charge
Reverse-phase column
Hydrophobicity
Can be automated
Double sort
Allowing direct elution from the second column into the MS
Extracted ion chromatograms(XICs)
How can we use MS to identify proteins
Many proteins are too big to identify by MS
Enzymatic proteolysis gives peptide fragments
Trypsin is cheap and reliable
Cleaves c-terminal to Arginine(R ) and Lysine (K)
Except when Arg (R ) is N terminal to Proline (P)
Exact peptide size includes abundance of natural isotopes
3-4 decimal places is needed in m/z
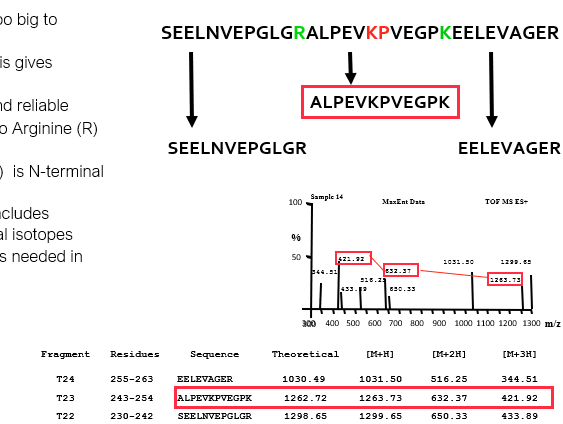
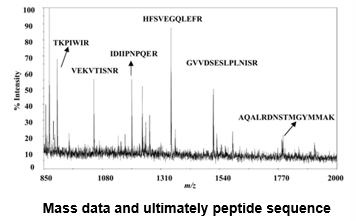
Identification of proteins from peptide m/z ratios
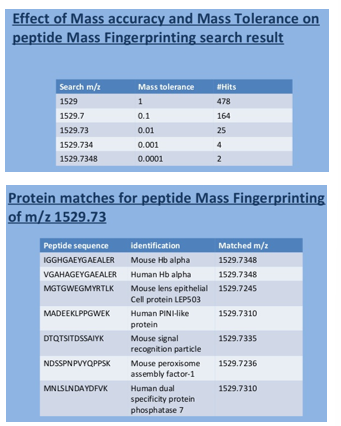
Databases exist containing the theoretical cleavage fragments of all known proteins with trypsin and other enzymes
Accurate peptide m/z ratio is present for each peptide in DB
Comparison of your peptide(s) gives a list of possible matches per peptide
Less hits with more accurate m/z ratios
Multiple peptides giving identity with the same protein support good quality identification
Problems were few peptide m/z's
Problems with common motifs
Found in multiple proteins
Sequencing peptides by MS-MS
Pick one peptide seen by MS at a time
Select for that peptide using quadrupole filter/ion trap
Collide it with inert gas
Peptide bond breaks
Fragments are varies proportions of the whole peptide
Analyse the fragments by TOF mass analysis
Use amino acid masses to calculate sequence
2 ion series
Y series (charge on C terminus) and
B series(charge on N terminus)
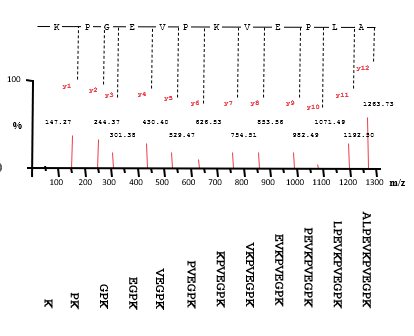
Y series ions shown
Identification of proteins from peptide sequence
Databases exist containing the 'theoretical' cleavage fragments of all known proteins with trypsin(and other enzymes)
Can examine all databases for possible coding sequence matches
Number of hits decreases and confidence increases with added peptide sequences of same protein
Problems where only one peptide sequenced
Problems with PTMs
Quantitative proteomics
MS is inherently non-quantitative
Quantitative analysis by multiple methods
Stable isotope labelling by amino acids in cell culture(SILAC)
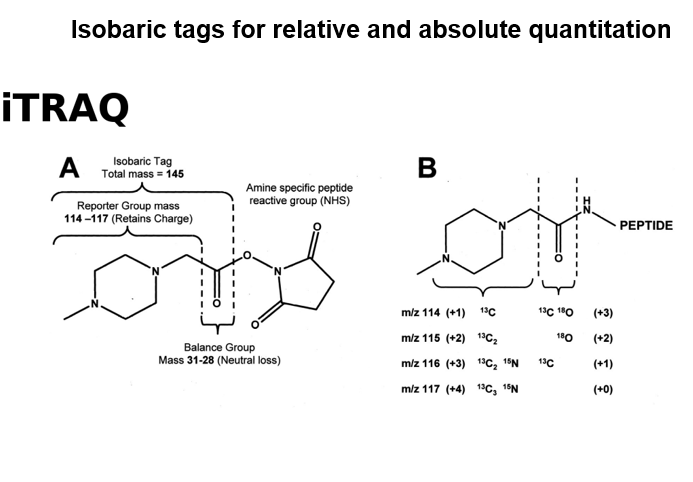
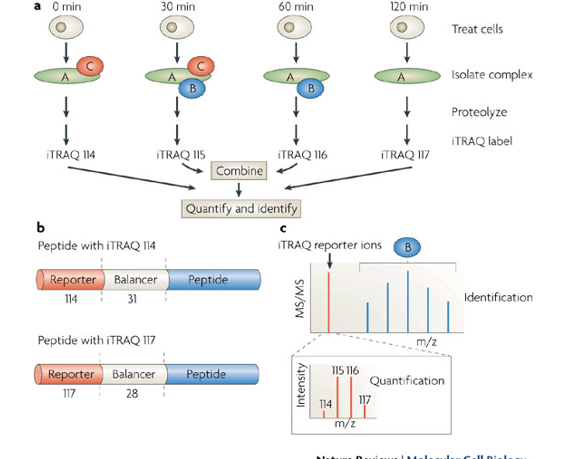
Isobaric tags for relative and absolute quantitation(iTRAQ)
Isobaric(same weight) - nominally same mass
iTRAQ as an example
Applications and the future
Protein interactions
Mapping of entire cellular systems
Drug effects
Disease biomarkers
Rapid diagnostics
From breath or other sample
Modelling biological responses
Improved specificity drugs
Improved understanding off complex diseases