Platelet Disorders
Platelet Disorders
Learning Objectives
Define thrombocytopenia and thrombocytosis.
Identify common causes of thrombocytopenia.
Recognize clinical manifestations of platelet disorders.
Classify qualitative vs. quantitative platelet defects.
Distinguish primary vs. secondary thrombocytosis.
Platelet Counts
Normal range: 150,000 - 450,000/μL.
Thrombocytopenia: <150,000/μL.
Thrombocytosis: >450,000/μL.
Lifespan: 7-10 days; 1/3 sequestered in spleen.
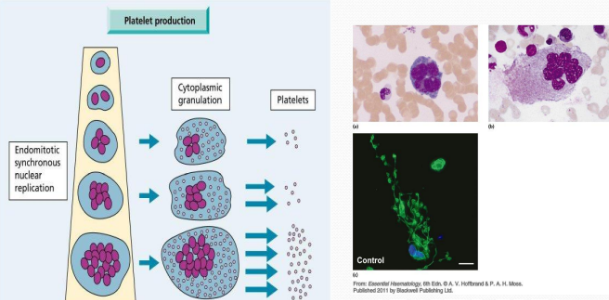
Platelet Production
Megakaryocytes undergo endomitotic synchronous nuclear replication.
Platelet production involves cytoplasmic granulation.
Platelet production occurs primarily in the bone marrow, where megakaryocytes differentiate and release platelets into the bloodstream. This process is regulated by various cytokines, such as thrombopoietin, which stimulate the proliferation and maturation of megakaryocytes.
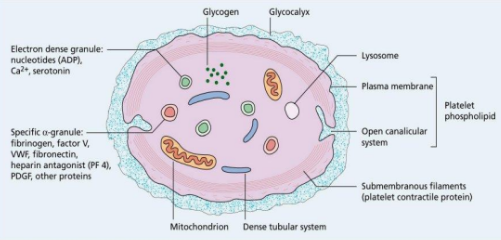
Structure of Platelet
Electron dense granules: Contain nucleotides (ADP), Ca2+, serotonin.
Specific α-granules: Contain fibrinogen, factor V, VWF, fibronectin, heparin antagonist (PF4), PDGF, and other proteins.
Other components: Glycogen, glycocalyx, mitochondrion, dense tubular system, lysosome, plasma membrane, platelet phospholipid, open canalicular system, submembranous filaments (platelet contractile protein).
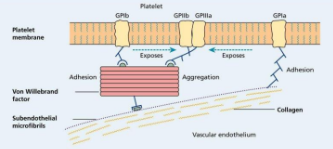
Function of Platelet
Adhesion: Platelets adhere to subendothelial microfibrils via von Willebrand factor (VWF) and GPIb.Aggregation: Upon activation, platelets aggregate through fibrinogen binding to GPIIb/IIIa receptors, leading to the formation of a platelet plug at the site of vascular injury.
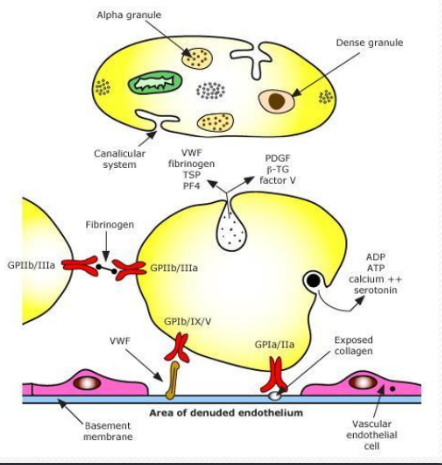
Aggregation: Platelets aggregate via GPIIb/IIIa, fibrinogen, and other factors.
Vascular endothelium is involved in the process.
Key components: Collagen, VWF, fibrinogen, TSP, PF4, PDGF, β-TG, factor V, ADP, ATP, calcium ++, serotonin, GPIb/IX/V, GPIa/IIa.
Manifestations of Platelet Disorders
Quantitative and Qualitative
Platelet/Vessel Wall Diseases: Mucosal bleeding and petechiae are common; deep hematomas are rare; bleeding from skin cuts is persistent; equal sex distribution.
Coagulation Diseases: Mucosal bleeding and petechiae are rare; deep hematomas are characteristic; bleeding from skin cuts is minimal; >80% male.
Platelet Disorders
Platelet disorders are the most common cause of bleeding.
Disorders can involve:
Decreased number (Thrombocytopenia)
Increased number (Thrombocytosis)
Defective function
Classification of Platelet Disorders
Quantitative Disorders
Thrombocytopenia
Decreased production
Increased destruction
Abnormal distribution
Thrombocytosis
Qualitative Disorders
Inherited disorders (rare)
Acquired disorders
Medications (e.g., ASA)
Chronic renal failure
Cardiopulmonary bypass
Causes of Thrombocytopenia
Decreased Production
Selective megakaryocyte depression
Congenital
Acquired (drug, chemical, viral)
Part of general bone marrow failure
Cytotoxic drugs and radiotherapy
Aplastic anemia
Marrow infiltration (by malignancy)
Megaloblastic anemia
HIV infection
Increased Consumption of Platelets
Immune
Autoimmune
Idiopathic (ITP; “Idiopathic Thrombocytopenic Purpura” – old name)
SLE, Lymphoproliferative disorders (CLL, Lymphoma)
Disseminated intravascular coagulation
Thrombotic thrombocytopenic purpura
/
/
Distribution
Pseudothrombocytopenia (Platelet clumps)
Splenomegaly
Dilutional (Massive transfusion)
Splenic pool: 30% normally vs. 60-90% in splenomegaly
Circulating platelets: 70% normally vs. 10-40% in splenomegaly
Approach to the Thrombocytopenic Patient
History:
Is the patient bleeding?
Are there symptoms of a secondary illness? (neoplasm, infection, autoimmune disease)
Is there a history of medications, alcohol use, or recent transfusion?
Are there risk factors for HIV infection?
Is there a family history of thrombocytopenia?
Do the sites of bleeding suggest a platelet defect?
Assess the number and function of platelets:
CBC with peripheral smear
Platelet function study (PFA and platelet aggregation studies)
vWD screen
ITP (Immune Thrombocytopenia)
Immune-mediated acquired disease of adults and children
Characterized by:
Low platelet count (<100 x /L, transient or persistent)
Increased risk of bleeding due to impaired clotting mechanism
Currently no definitive diagnostic criteria exist for primary ITP
Considered a diagnosis of exclusion
Diagnostic Approach in Suspected ITP
To exclude other causes of thrombocytopenia, recent updates recommended basic evaluation should consist of:
Patient history – necessary to rule out other causes of thrombocytopenia
Physical examination – normal except for signs of thrombocytopenia; no adenopathy or splenomegaly
Complete blood count showing isolated thrombocytopenia with large platelets
Clinical or laboratory evidence for other causes of thrombocytopenia
Proposed Mechanism of Immune Dysregulation in ITP
T cells are activated upon recognition of platelet-specific antigens on the APCs and therefore induce antigen-specific expansion of B cells.
Clinical Manifestations of Platelet Disorders
Skin purpura, superficial bruising, epistaxis, menorrhagia.
Mucosal hemorrhage is seen in severe cases and intra-cranial hemorrhage is rare.
Thrombotic Thrombocytopenic Purpura (TTP)
Pathogenesis
Von Willebrand factor (VWF) consists of a series of VWF multimers each of molecular weight (MW) 250 kDa which are covalently linked.
Under physiological circumstances a metalloprotease ADAMTS13 cleaves high molecular weight multimers at a Tyr-842-Met-843 bond and the resulting VWF has an MW of 500-20 000 kDa.
In non-familial TTP, an antibody develops to the metalloprotease and so blocks cleavage of VWF multimers.
In congenital form of TTP, the protease appears to be absent.
In both cases, the resultant ultra-large VWF multimers can bind platelets under high shear stress conditions and lead to platelet aggregation.
Disseminated Intravascular Coagulation (DIC)
Pathogenesis
Widespread activation of coagulation.
Endothelial damage leads to:
Microthrombi in the circulation
↓ Platelets due to generalized platelet aggregation
Fibrinolysis + FDPs
Defective Platelets Function
A defect in platelet function is suspected if there is prolonged bleeding time with or without skin or mucosal hemorrhage in the presence of a normal platelet count.
Clinical and Laboratory Findings in Hemophilia A, Factor IX Deficiency, and von Willebrand Disease
| Feature | Hemophilia A | Factor IX Deficiency | von Willebrand Disease | |---------------------------|--------------|-----------------------|------------------------|
| Inheritance | Sex-linked | Sex-linked | Dominant (incomplete) |
| Main sites of hemorrhage | Muscle, joints, post-trauma or postoperative | Muscle, joints, post-trauma or postoperative | Mucous membranes, skin cuts, post-trauma or postoperative |
| Platelet count | Normal | Normal | Normal |
| PFA-100 | Normal | Normal | Prolonged |
| Prothrombin time | Normal | Normal | Normal |
| Partial thromboplastin time | Prolonged | Prolonged | Prolonged or normal |
| Factor VIII | Low | Normal | Normal |
| Factor IX | Normal | Low | Normal |
| VWF | Normal | Normal | May be moderately reduced |
| Ristocetin-induced platelet aggregation | Normal | Normal | Impaired |
Classification of von Willebrand Disease
Type | Description |
|---|---|
1 | Quantitative partial deficiency |
2 | Functional abnormality |
3 | Complete deficiency |
Type 2 VWD Subtypes
Subtype | Platelet-associated function | Factor VIII binding capacity | High MW VWF multimers |
|---|---|---|---|
2A | Decreased | Normal | Absent |
2B | Increased affinity for GPIb | Normal | Usually reduced/absent |
2M | Decreased | Normal | Normal |
2N | Normal | Reduced | Normal |
Thrombocytosis
Increased platelet counts can be due to a number of disease processes:
Essential (primary)
Essential thrombocytosis (a form of myeloproliferative disease)
Other myeloproliferative disorders such as:
chronic myelogenous leukemia,
polycythemia vera,
myelofibrosis
Reactive (secondary)
Inflammation
Surgery (which leads to an inflammatory state)
Hyposplenism (decreased breakdown due to decreased function of the spleen)
Hemorrhage and/or iron deficiency