Ageing
Aged cells exhibit many different properties to younger cells eg:
slower division
decreased cellular metabolism
change in cellular function eg production of non-physiological protein, cytokines and growth factors
can’t apoptose
decreased receptor sensitivity
reduced withstanding ROS
DNA instability
There are many different theories as to why we/our cells die eg
programmed to die - shouldn’t live after reproducing due to competition
Hayflick limit of cell division
Telomere shortening
IGF-1 signalling via FOXO TF deficits with age
reduced immunity over time
Hayflick limit: Is replicative senescence the cause of aging?
fibroblasts isolated and cultured from human tissue
placed in culture vessel with nutrient medium
divide and form confluent layer on vessel surface
half of cells discarded, remainder grow to confluency - 1 passage
repeat above step
replication slows and stops after 50 ± 10 passages (Hayflick limit, replicative senescence)
Telomere shortening:
What are telomeres? Telomeres are repetitive DNA sequences found at the ends of all human chromosomes to protect them from damage. Telomeres contain thousands of TTAGGG repeats and gradually shorten as cells divide.
Telomerase is a ribonucleoprotein with two main components: telomerase reverse transcriptase (hTERT) which contains the catalytic component, and hTR RNA aka TERC which acts as a docking site to recognise telomeres and acts as a template for telomere synthesis.
Telomerase adds telomere repeats to the ends of chromosome and is not expressed in most differentiated somatic cells, but is expressed in stem cells and some cancer cells.
Reversing ageing using telomerase may increase tumour risk. Most mouse tissues also have an active telomerase but this doesn’t have any effect on mortality/reducing aging.
It has been proven that viral delivery of hTERT to mice causes telomere expansion and increases health span, however this mouse model was genetically resistant to cancer and it worked better on older mice.
Mice with very long telomeres present with less metabolic aging and longer lifespan.
Another theory is the ‘wear and tear’ theory which is the metabolic ‘rate of living’/burn-out’. ‘Duration of life varies inversely as the rate of energy expenditure.’ Affected by nutrients/diet, mitochondrial dysfunction, free radical generation and oxidative stress/accumulated cellular and DNA damage.
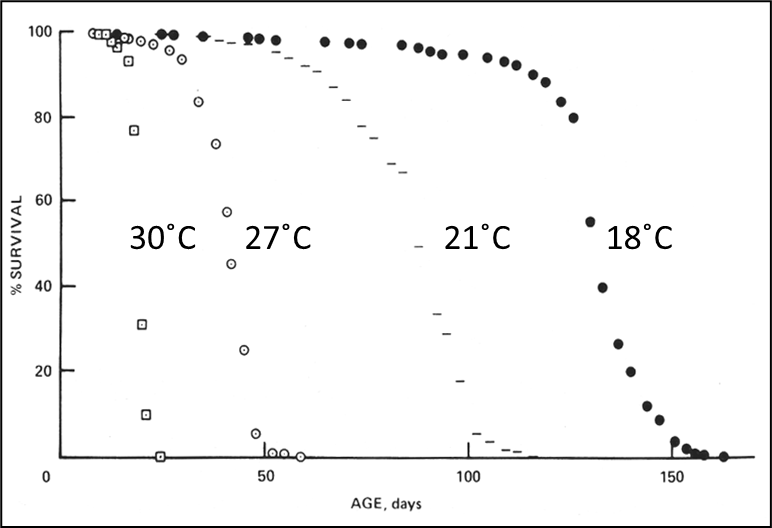
Effect of temperature on Drosophila population. For every change in approximately 10 degrees C, 2-3 fold change in lifespan.
Nutrients/diet and aging:
Essential amino acid reduction in food increases lifespan in yeast, flies and mice. This is mediated by TOR (target of rapamycin) signalling. Autophagy increases and cell growth and protein synthesis decrease, extending lifespan. Rapamycin has been shown to increase lifespan.
Drosophila is often used as an ageing model due to its short lifespan meaning the entire lifespan can be studied quickly. Median lifespan of 50 days with a max of 90 days. Gene mutants affect longevity and it was found that caloric restriction increases healthspan.
One study in which old and young mice had their circulatory systems surgically fused together showed a reversal in age related cardiac hypertrophy compared with isochronic old pairs (old-old fused) due to circulating growth differentiation factor 11. However rather than ‘deaging’ this may have just been a case of rejuvenating stem cell niches. Alongside GDF11, CCL11 and oxytocin have also been suggested as causative factors.
Progeroid syndromes are diseases of premature aging.
Werner’s syndrome was first described in 1904 and affects 1 in 1,000,000. Rate in Japan is 1 in 40,000. Manifests in adulthood with the first clinical sign being lack of growth spurt in puberty. Werner patients are on average 13cm shorter and 20kg lighter than the general population. From age of 30 the disease is progressive and death occurs in 40s, with heart disease being a major contributor.
Clinical features of Werner’s:
Cataracts (bilateral)
Dermatological pathology (tight, atrophic skin, pigmentary alterations, ulceration, hyperkeratosis, etc)
Short stature
Premature greying, thinning of scalp hair
Hypogonadism
Neoplasms (rare sarcomas)
Abnormal voice (high-pitches, squeaky or hoarse)
Type 2 Diabetes mellitus
Osteoporosis
Atherosclerosis (history of myocardial infarction)
Werner syndrome is an autosomal recessive condition in which the WRN gene is affected, which encodes RecQL2 helicase protein, 3-5’ exonuclease and is responsible for 3’-5’ helicase activity. WRN’s absence leads to abnormal DNA repair, replication and telomere maintenance.
Mutations in WRN can occur in many different places along the gene and may be nonsense (changes AA to stop codon), insertion/deletion (causing frameshift)., substitution at splice (skipping of exons and frameshift). One case is due to missense mutations causing codon change affecting protein stability. Most mutations generate a truncated WRN which lacks NLS, found at the C terminal portion.
How do WRN cells differ from WT cells?
accelerated aging
premature replicative senescence - Hayflick limit = 20
prolonged S phase
accumulate toxic DNA intermediates → DNA damage and apoptosis
increased chromosomal instability
apoptosis reduced
How do WRN telomeres differ from WT telomeres?
issue with telomere maintenance
on average erode at similar rates to WT
telomere erosion not uniform?
may be sensitive to few dysfunctional telomeres
1 dysfunctional telomere could be enough to send cell into replicative senescence
WRN also binds to the C terminus of p53 in vivo. WRN fibroblasts show reduced p53 mediated apoptosis and there is increased cancer cases due to reduced suppression of genomic instability. WRN/p53 double KO mice show further increased rate of tumour development and mortality.
Wrn knockout mice didn’t display obvious premature ageing/spontaneous cancer predisposition. Murine Wrn may be functionally redundant with other helicases compensating. However a combination of Wrn and Terc mutations does show a phenotype similar to progeria.
Differences between Werner syndrome and normal ageing:
no increased tendency for neurodegeneration (may be due to lack of time for this to occur)
no prostate problems in men
no immune changes
Hutchinson-Gilford syndrome:
first described in 1886, classified as progeria in 1904
1 in 8,000,000
both autosomal dominant and autosomal recessive
affects young people
Clinical features:
First signs in 1st year, slow growth, dwarfism
Lack of hair
Disproportionately large head
’Pinched' facial features
Lipodystrophy (almost complete absence of subcutaneous fat).
Incomplete extension at the knees and elbows - stiff joints
Coronary artery disease
Generally a senile appearance
By age 10 patients hair start turning grey
Individuals often die in their teens typically of heart disease
Difference between progeria and typical aging:
no prostate problems
no increased cancer/cataracts risk
rapid atherosclerosis development, high blood pressure rare
diabetes rare
don’t suffer mental degeneration/dementia etc
Progeria caused by mutation to lamin A gene (LMNA). Lamins are structural protein components of the nuclear lamina, a protein network underlying the inner nuclear membrane which determines nuclear shape and size. Lamins = intermediate filament class.
Many progeria cases are caused by the same de novo single-base substitution from C to T causing silent Gly to Gly change at codon 608, exon 11. Activates a cryptic splice site so the protein product has a 50 AA deletion near the C terminus - aberrant progerin protein.
Lamin functions:
mitosis
DNA synthesis/repair
RNA transcription/processing
apoptosis
chromatin organisation
gene expression regulation
It is likely that progerin has distinct functions.
Progeria cell dysfunction:
reduced lifespan in culture
irregular nuclear phenotypes eg blebbing (blister-like protrusions) of nuclear envelope
altered chromatin organisation
reduced telomere length
chronic DNA damage response
hTERT extends progerial cell lifespan by preventing entry into senescence, and also rescues DNA-damage/proliferative defects associated with progerin.