Pharmacokinetics
Overview of Pharmacokinetics
Definition: The relationship between drug dose and plasma concentration over time.
What does the body do to the drug?
Four Main Components:
Absorption
Distribution
Metabolism (Biotransformation)
Elimination
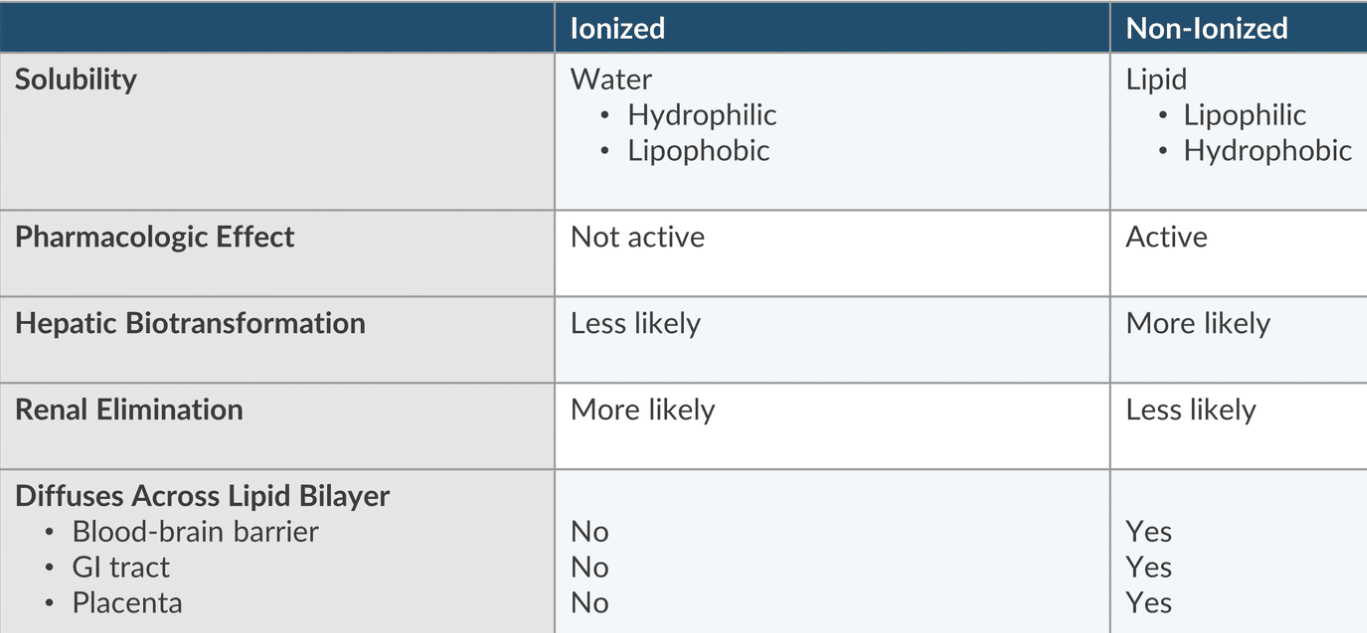
Acid-Base
An acid is a substance that donates a proton: HA H+ + A-
a base is a subsance that accepts a proton: B + H+ BH+
Ionization is dependent on two Factors:
The pH of the solution
The pKa of the drug
Ion Trapping
Only un-ionized, lipophilic drugs diffuse across membranes. Upon crossing, they ionize based on local and , resulting in varying drug concentrations and ionization levels on each side of the membrane.
After it does, it will ionize based on pKa and pH of the solution on this side of the membrance.
Therefore, the drug concentration and the degree of ionization will differ on each side of the membrane.
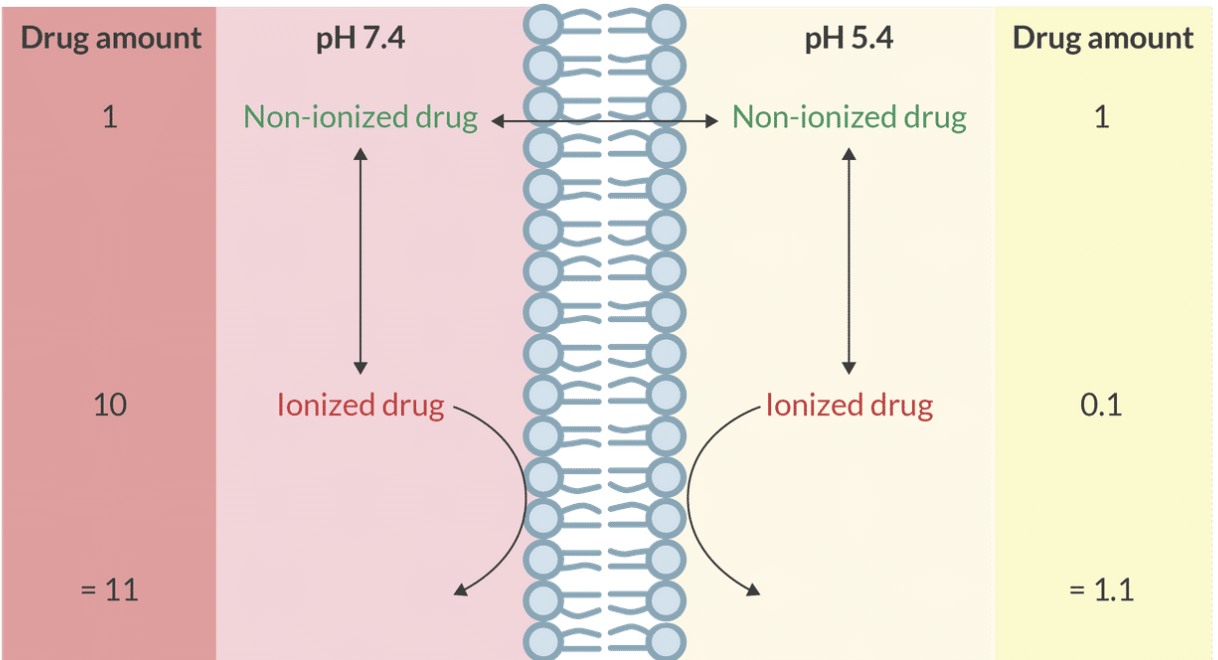
Fetal Ion Trapping
Mechanism: Un-ionized drugs cross the placenta and become ionized in the relatively acidic fetal environment. Once ionized, they cannot diffuse back, leading to drug accumulation in the fetus.
Key Factors:
Maternal alkalosis increases the risk.
Lidocaine is highly susceptible to trapping.
Chloroprocaine is least likely to be trapped due to its high pKa and rapid metabolism in maternal blood.
Absorption
Definition: The process of drug movement from the site of administration to the bloodstream (plasma).
Bioavailability:
Definition: The fraction of the drug dose that reaches systemic circulation.
Example: Intravenous (IV) administration has a bioavailability of 1 (or 100%).
Factors Affecting Absorption:
Un-ionized drugs absorb better than ionized drugs.
Acidic drugs are better absorbed in the acidic stomach, while basic drugs are absorbed in the alkaline intestine.
First-Pass Metabolism:
Venous drainage from stomach and small intestines flows to the liver, leading to first-pass metabolism.
Venous drainage from the mouth and esophagus goes to the superior vena cava, bypassing the liver.
Rectal administration partially bypasses the portal system, making it a useful option for children or individuals who cannot take oral medications.
Note: Rectal administration can be erratic and may cause irritation to the rectal mucosa.
Transdermal Administration:
Provides extended release.
The stratum corneum (first sub-layer of the epidermis) is a barrier to most medications except lipid-soluble drugs (e.g., nitroglycerin, scopolamine, fentanyl, EMLA, clonidine).
Subcutaneous Absorption:
Relies on diffusion from the injection site to the bloodstream.
Diffusion rate is affected by blood flow to the injected tissue and the drug formulation (solutions [already ready] are absorbed faster than suspensions[must be broken down first before being absorbed]).
IV administration bypasses absorption entirely, avoiding first-pass metabolism.
Distribution
Definition: After absorption, drugs are distributed throughout the body via the bloodstream.
Distribution Characteristics:
The vessel-rich group (brain, heart, liver, kidneys) receives a greater portion of cardiac output (75%) but constitutes only 10% of body mass.
This leads to faster equilibration with plasma concentration due to increased blood flow
The muscle and skin constitute 50% of body mass but receive only 19% of cardiac output.
Fat (20% body mass) receives 6% of cardiac output.
The vessel-poor group (bone, ligaments, cartilage) is 20% body mass and receives <1% of cardiac output.
Fat and skin have a larger capacity for storing lipophilic drugs, which leads to a larger reservoir after infusion or a large bolus dose.
Law of Mass Action:
When plasma concentration exceeds tissue concentration, drugs move down their concentration gradients (vice versa).
Volume Distribution (Vd):
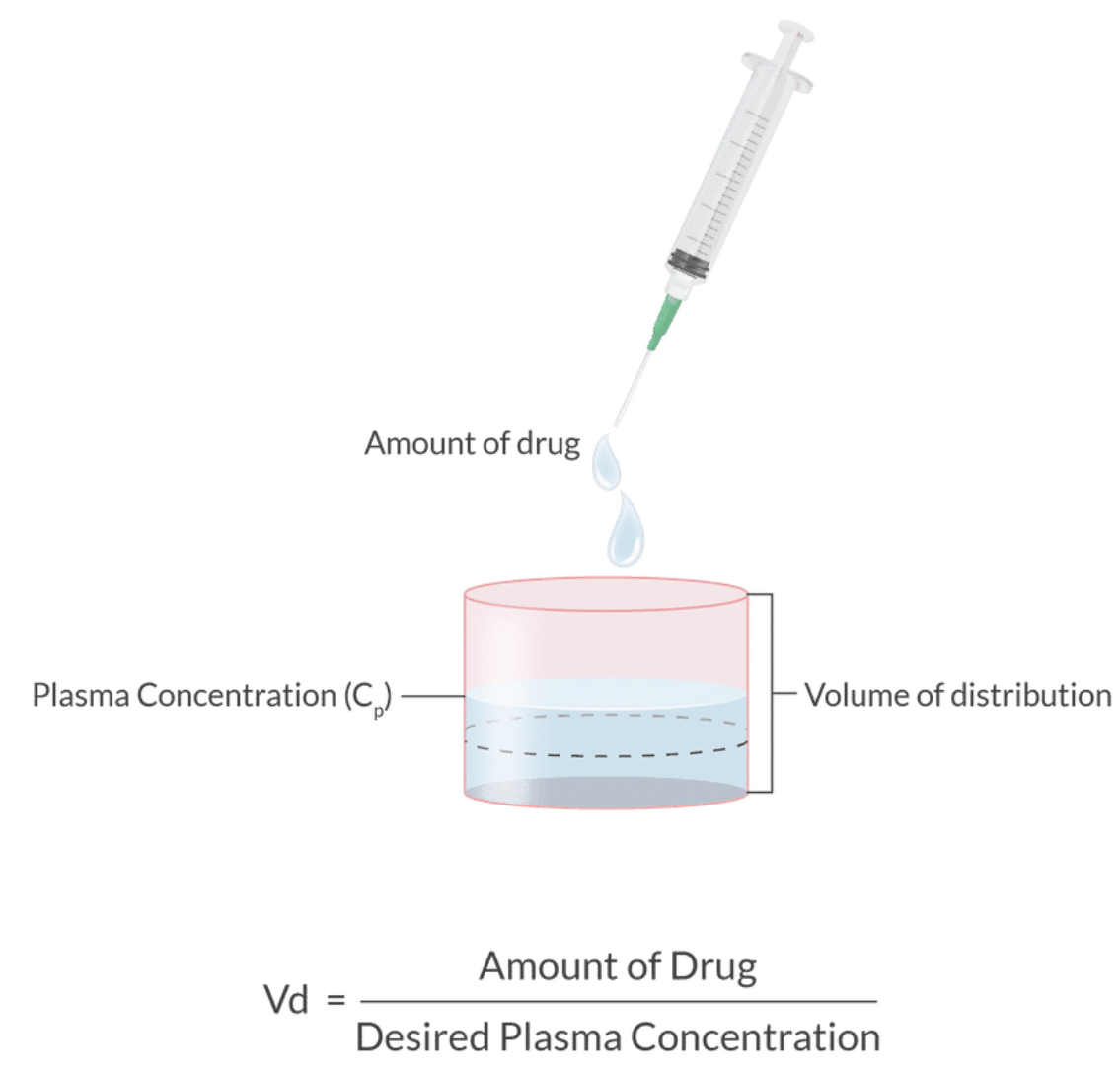
Definition: Relationship between drug dose and the resulting plasma concentration, indicating how much a drug spreads after entering the body.
Formula:
Characteristics:
Small volume distribution = drug remains primarily in the bloodstream.
Large volume distribution = drug disperses into tissue (fat/muscle).
Vd assumes two things:
1) drug distributes instanataneously
Full equilibration at time = 0
2) drugs aren’t subject to biotransformation or elimiation before it’s fully distributed.
Affecting Factors of Vd:
Drug characteristics: molecular size, ionization, protein binding.
Patient characteristics: pregnancy, burns, etc.
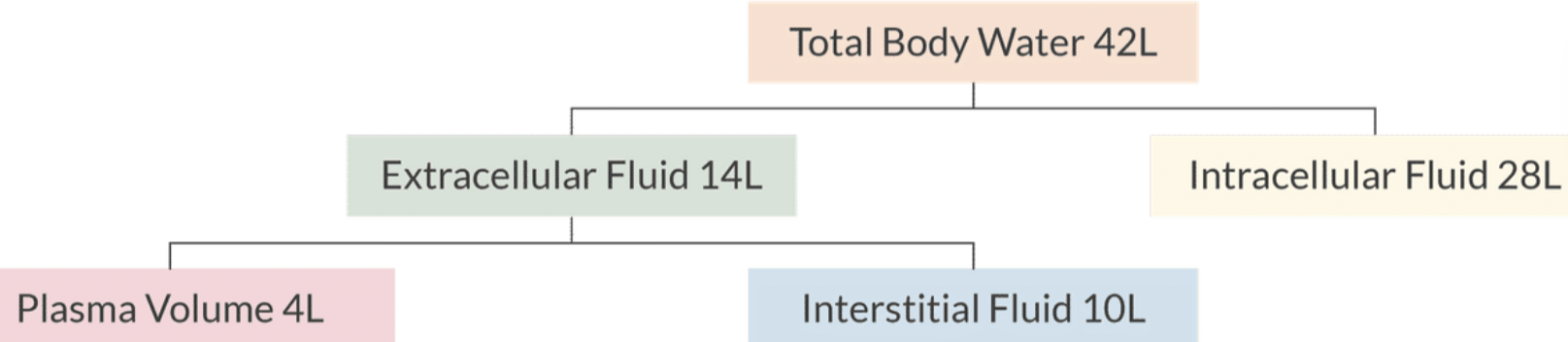
Body Water Distribution:
Total body water in a 70 kg male = 42 liters, subdivided into:
Intracellular fluid: 28 liters
Extracellular fluid: 14 liters (4 liters plasma volume, 10 liters interstitial fluid).
Lipophilic Drugs:
Volume distribution greater than total body water indicates higher lipophilicity, requiring increased doses.
Example: Propofol.
Hydrophilic Drugs:
Volume distribution less than total body water indicates higher hydrophilicity, allowing for reduced doses.
Example: Neuromuscular blockers.
Loading Dose:
Purpose: Achieve a predetermined plasma concentration.
Formula:
IV Anesthetics:
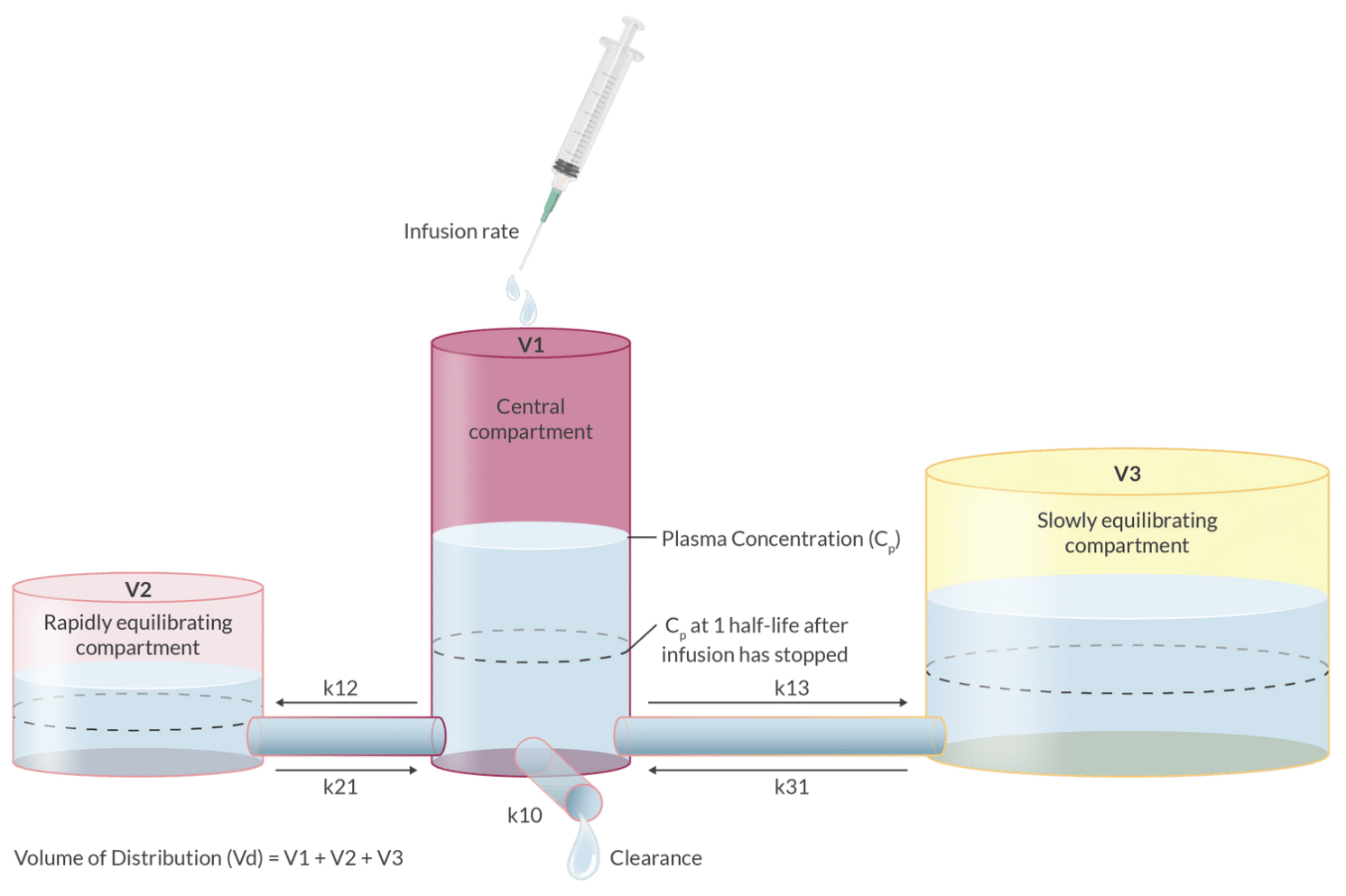
Display two compartments (central and peripheral).
Volume distribution at steady state involves both compartments.
The central compartment represents the plasma and highly perfused organs, while the peripheral compartment includes less perfused tissues. This multi-compartmental model helps in understanding the distribution kinetics of IV anesthetics and optimizing dosage regimens.
Compartment Models:
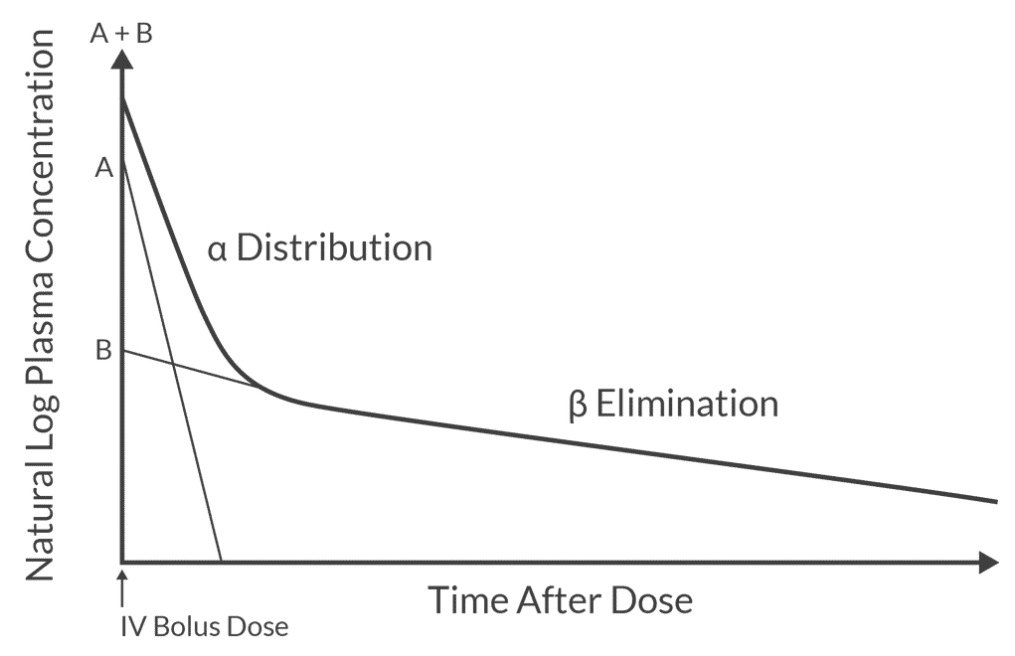
Two-compartment model shows biphasic decrease in plasma concentration after IV bolus (alpha distribution and beta elimination with differing slopes).
Three-compartment model expands this concept further, accounting for a third compartment, often representing slowly equilibrating tissues, which provides an even more accurate representation of the drug's pharmacokinetics in complex biological systems.
Elimination
Definition: The process of removing drugs from the body.
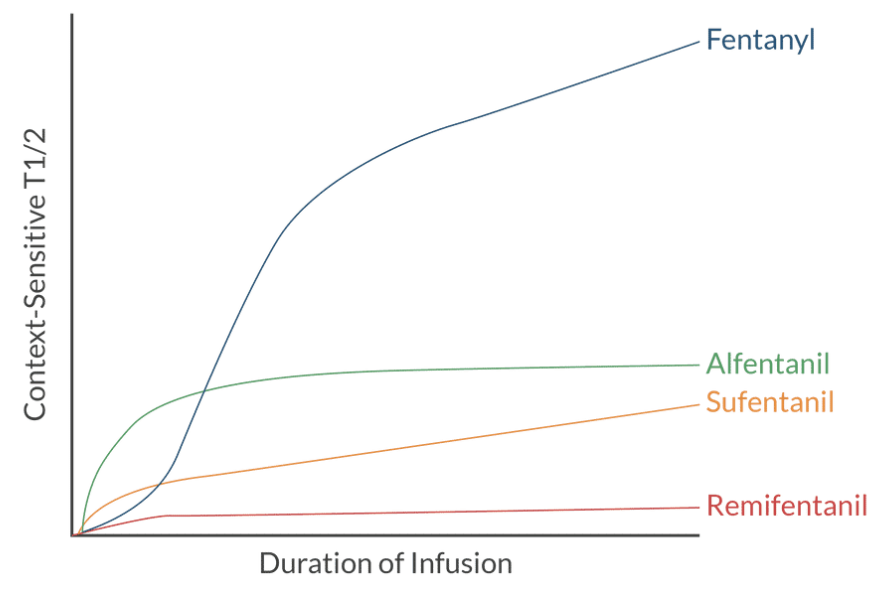
Contect-Sensitive Half-Life:
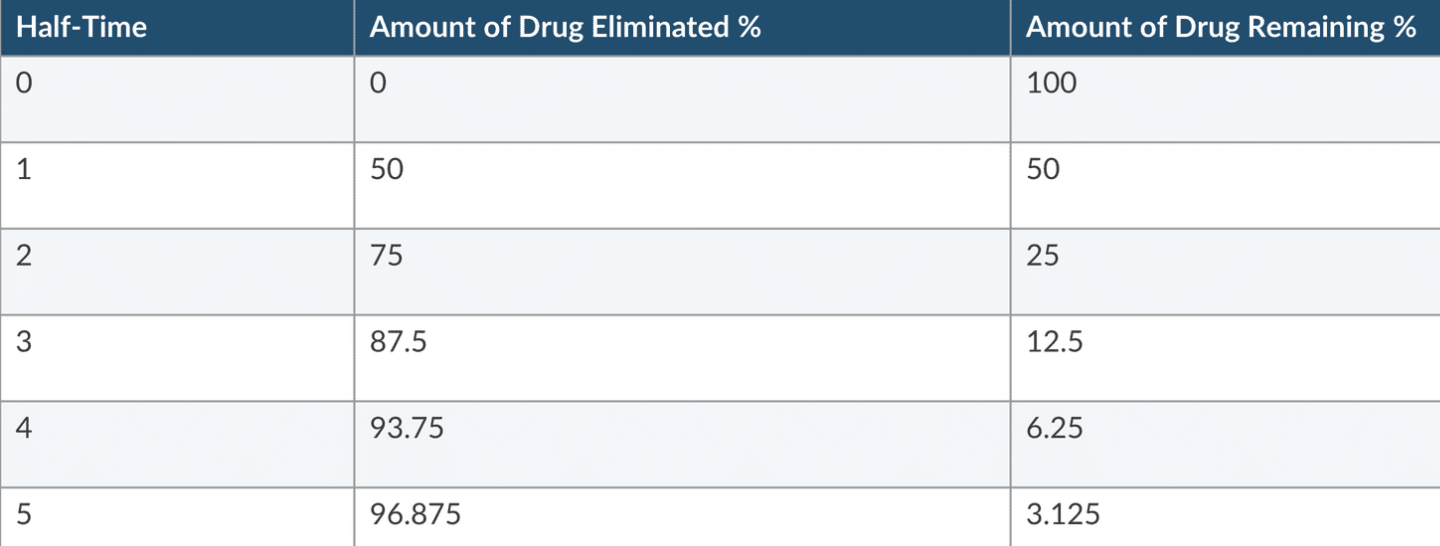
Definition: Time required for 50% of the drug to leave the body after a rapid IV injection. Measures a constant fraction and NOT a constant amount.
Different from elimination halftime (the time for 50% removal during the elimination phase, applicable to one-compartment models).
Context-Sensitive Half-Time:
Definition: Time needed for plasma concentration to decrease by 50% after cessation of infusion.
Longer infusions result in greater drug accumulation in peripheral compartments, increasing elimination half-time.
Metabolism (Biotransformation)
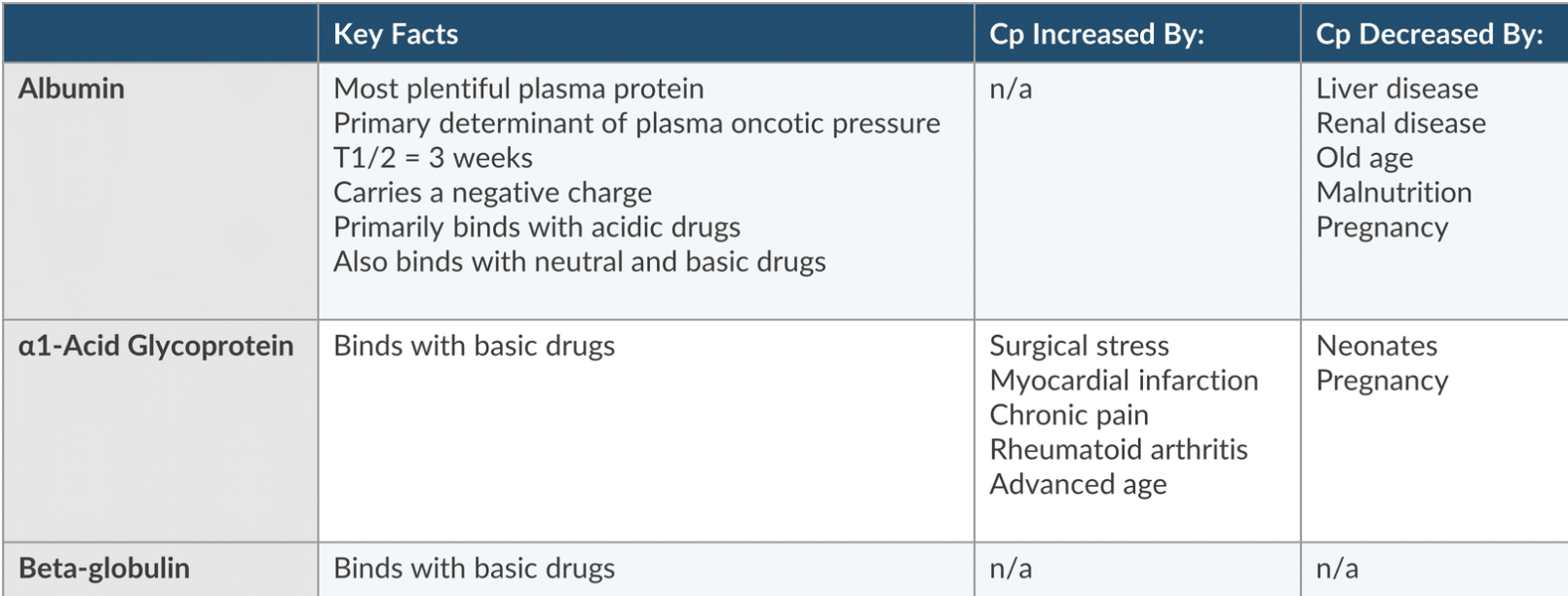
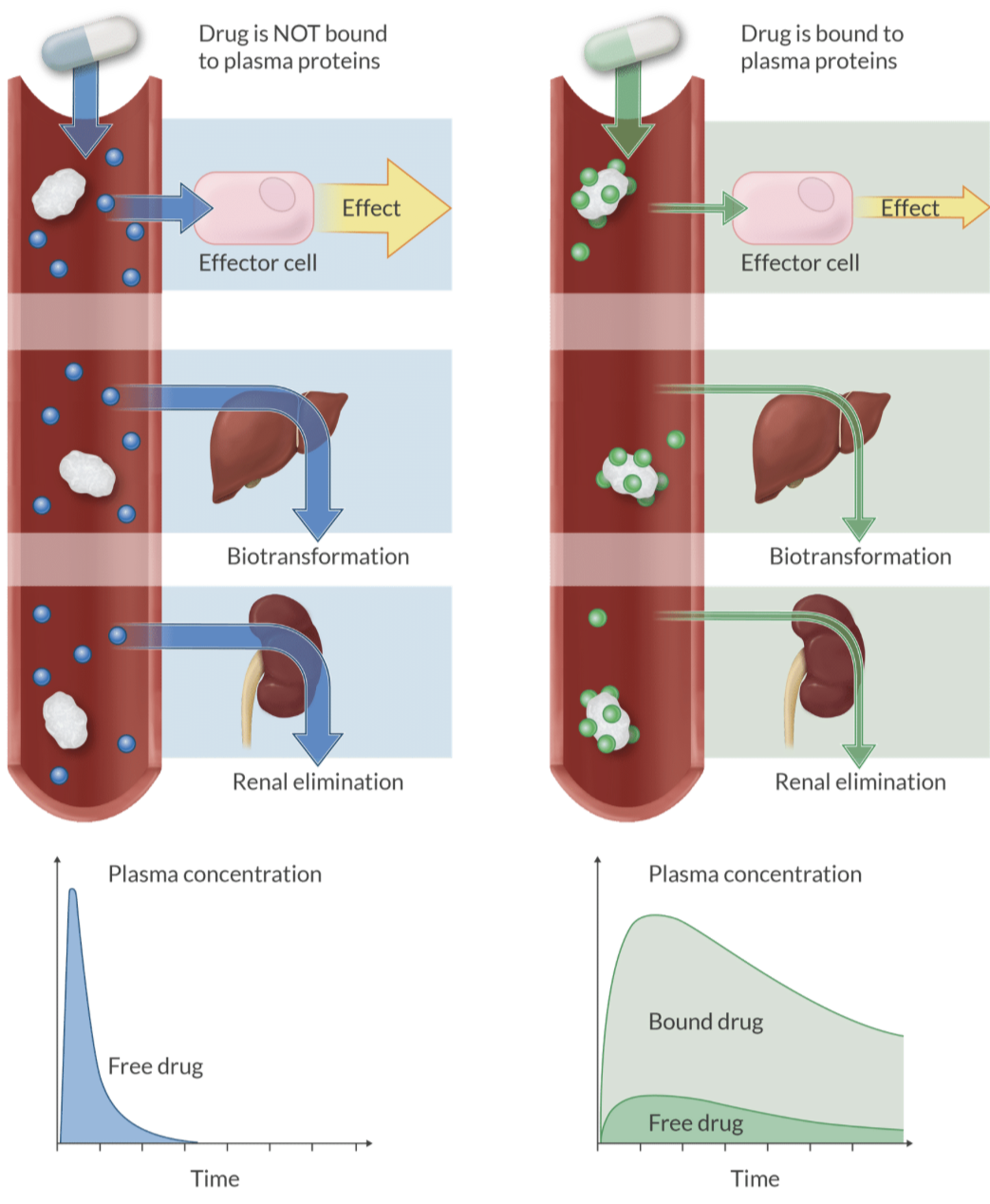
Definition: The process in which the liver synthesizes plasma proteins that bind to drugs, affecting their activity.
Plasma Proteins:
Albumin: the most plentiful protein that’s negatively charged and binds acidic drugs.
It determines oncotic pressure and has a T1/2 of 3 weeks
Plasma concentration is increased by liver and renal disease, old age, malnuitrition, and pregnancy
alpha-1 acid glycoprotein and beta globulin (binds basic drugs).
Alpha-1 acid glycoprotein: A positive acute phase protein and its levels are influenced by inflammation and stress.
Measuring Percent Change Formula:
Alteration in protein binding: cardiopulmonary Bypass, ECMO, bilirubin, and thyroxin
Factors Influencing rate Metabolism:
Blood flow to the metabolizing site.
Genetic factors and enzyme activity (inducers and inhibitors).
Kinetics:
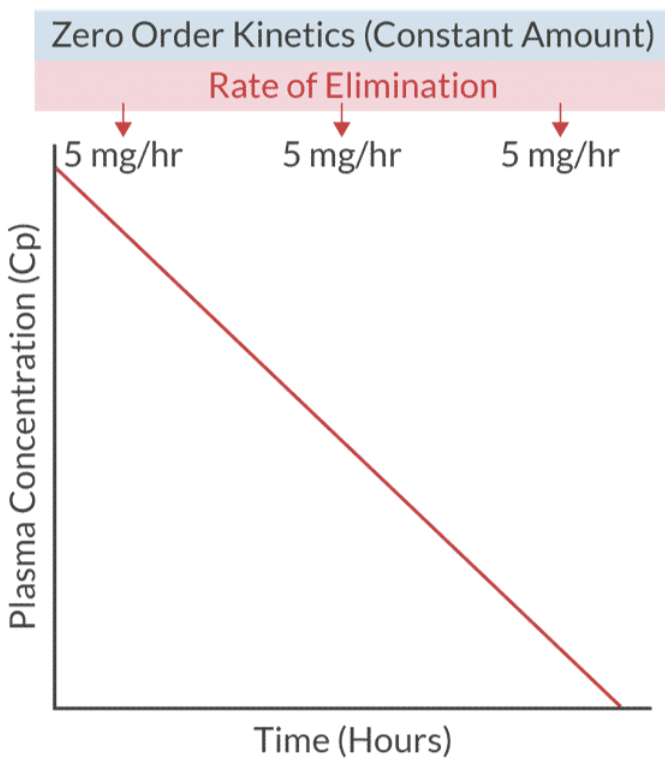
Zero Order Kinetics: Constant amount of drug metabolized per time (e.g., theophylline, heparin, Aspirin, Phenytoin, ETOH, and Warfrin).
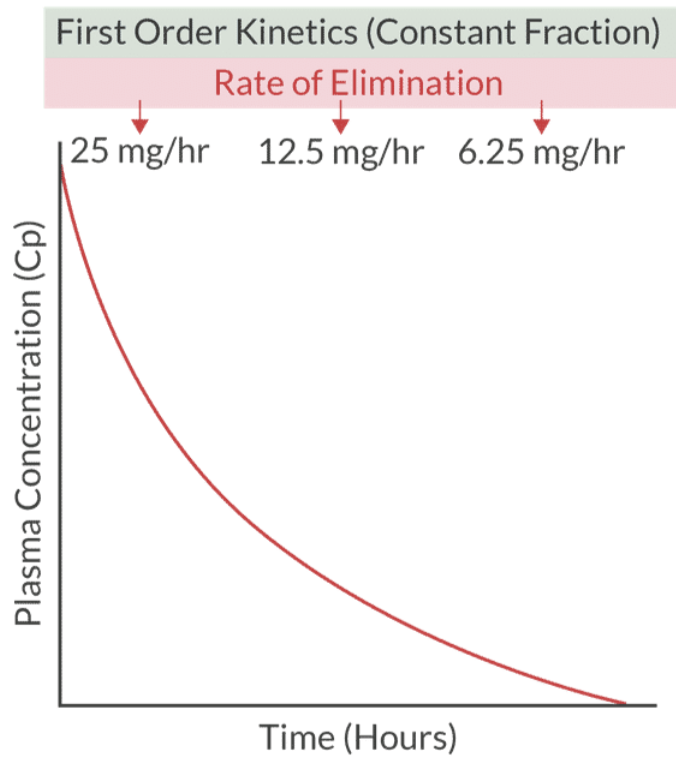
First Order Kinetics: A fraction of drug metabolized per time (e.g, most common used in clinical practice)
can convert to zero order if saturation occurs.
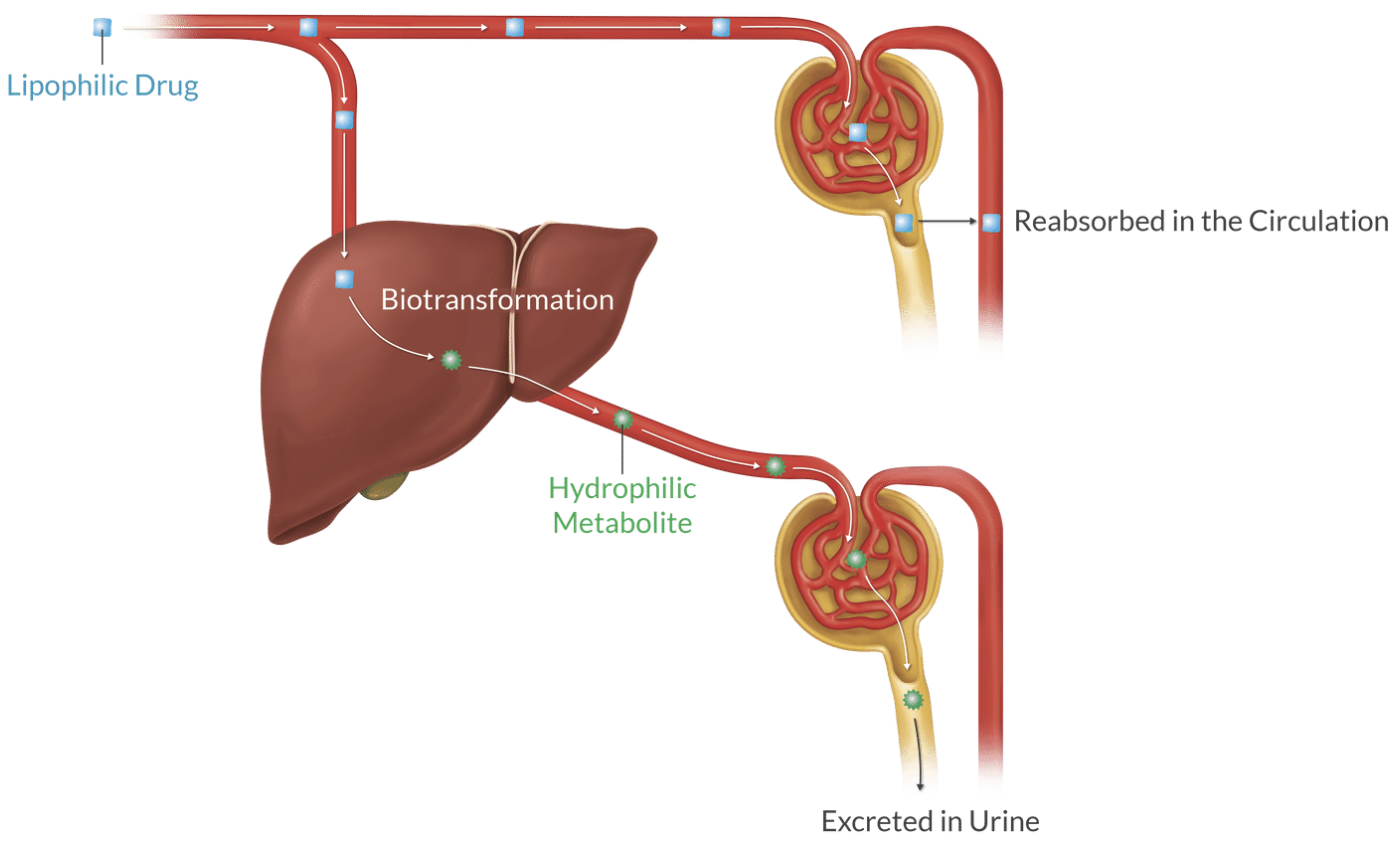
Sites of Metabolism:
Primarily in the liver (smooth endoplasmic reticulum), but also in kidneys, plasma (Hoffman and hydrolysis), lungs, and intestines.
Purpose of metabolism is to change lipid-soluble, active compounds into water-soluble, inactive byproducts (waste)
Metabolism Phases:
Phase I: Adjusts the compound for Phase II (modification)
Oxidation: removes electron
Reduction: adss electron
Hydrolysis: adds water to split it apart (e.g., esters)
Phase II: Involves conjugation to make molecules inactive and water-soluble for excretion.
Adds a highly polar, water-soluble substrate
glucuronic Acids, Glycine, Acetic acid, Sulfric acid, or a methyl group
Enterohepatic Circulation: excreted into the bile and it’s reactivated in the intestine which leads to reabsoprtion back into the systemic circulation (e.g., Warfarin and Diazepam).
Phase III: ATP-dependent carrier potein transportion of drugs across the cell membrane for elimination.
4 Key Metabolic Pathways:
Pseudocholinesterase: Drugs include succinylcholine, Mivacurium, and ester local anesthetics (cocaine also involves hepatic.
Nonspecific Esterases: Includes remifentanil, Esmolol (RBC esterases), Atracurium (also invloves hoffman), Clevidipine, Etomidate (also involves hepatic), and remimazolam.
Alkaline Phosphatase: Engages fospropofol (prodrug).
Hoffman Elimination: Affected by pH and temperature (e.g., atracurium and cisatracurium).

Important Enzyme Systems:
Located in the smooth endoplasmic reticulum of the hepatocytes.
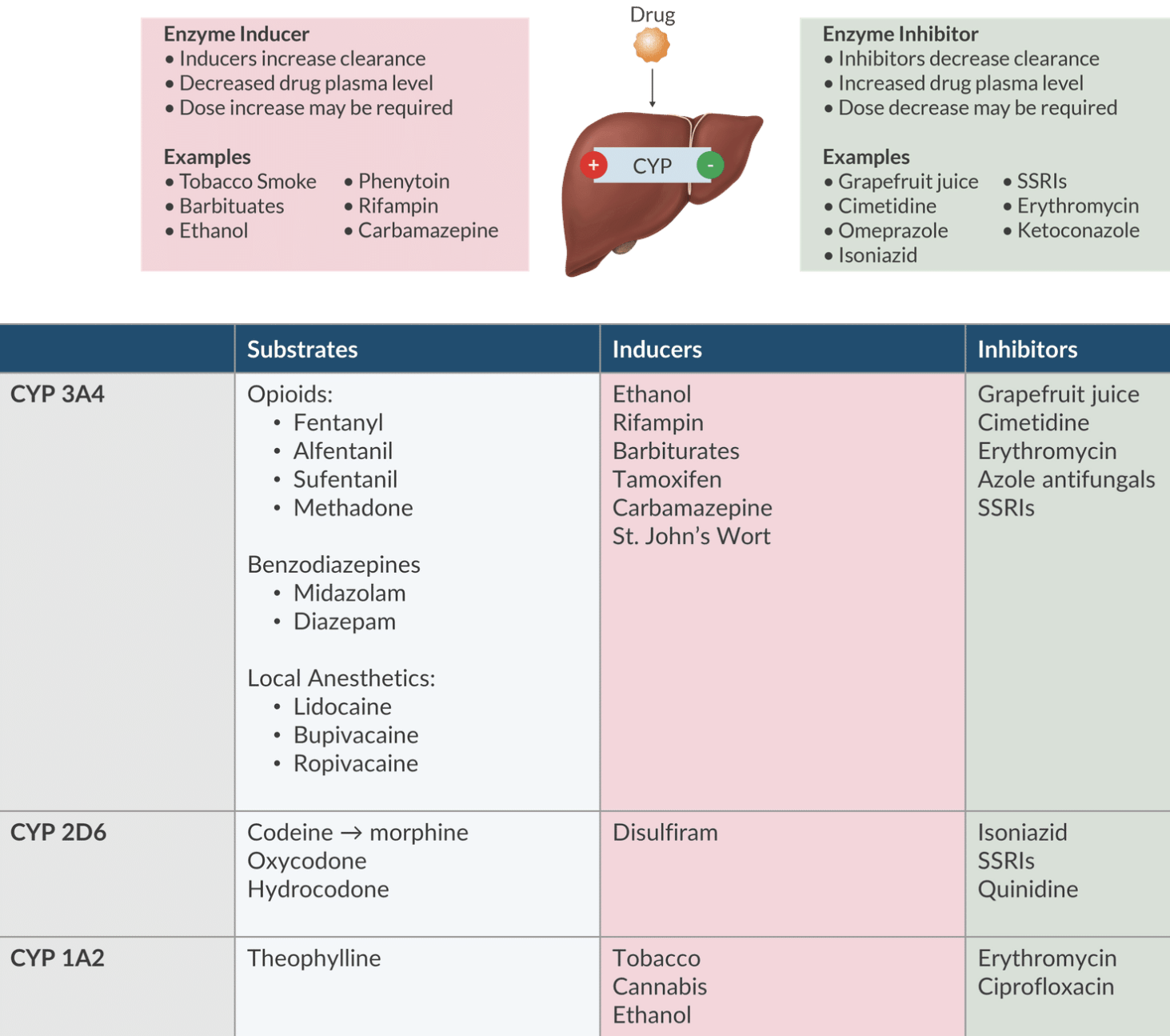
Cytochrome P450 System (CYP): CYP3A4 handles 50% of anesthetics.
Enzyme Inducers: Increase drug clearance (e.g., ethanol, rifampin, barbiturates, St. John’s Wort) - may require higher doses.
Enzyme Inhibitors: Decrease drug clearance (e.g., grapefruit juice, erythromycin, ciprofloxacin, and SSRIs) - may lower dose requirements.
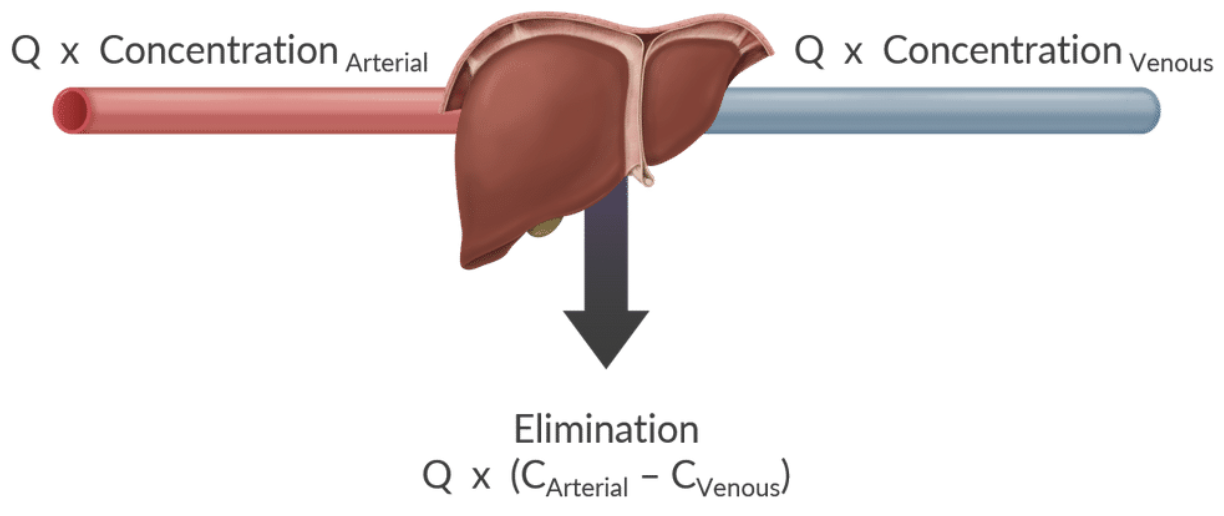
Hepatic Clearance
Definition: The rate at which drugs are cleared from the liver is influenced by blood flow and the hepatic extraction ratio.
Extraction Ratio:
Formula:
Effects: Extraction ratio of 1 means 100% drug removal.
Ratios >0.7 (high extraction) means clearance is dependent on liver blood flow (e.g., fentanyl, morphine, ketamine, and propofol)
Ratios <0.3 (low extraction) are dependent on liver extracting drug from blood (e.g., Rocuronium, diazepam, and methadone)

Renal Excretion
Definition: The elimination of drugs from the body through kidney excretion.
Mechanisms:
Unbound, small drugs pass freely from plasma to glomerular filtrate.
Unionized drugs are reabsorbed in the renal tubules; ionized drugs remain excreted based on urinary pH.
Renal elimination of drugs depends on urinary pH and polarity
Transport Systems:
Organic Anion Transporters for drugs like furosemide, thiazide, and pencillin
Organic Cation Transporters for drugs like morphine, meperidine, and dopamine
These proteins are located in the proximal renal tubules and secrete both acidic and basic compounds.
Metabolites may convert back to parent drugs (example: lorazepam).