3. Glucose uptake and storage 3_2024_MPAL_
Glucose Uptake and Storage
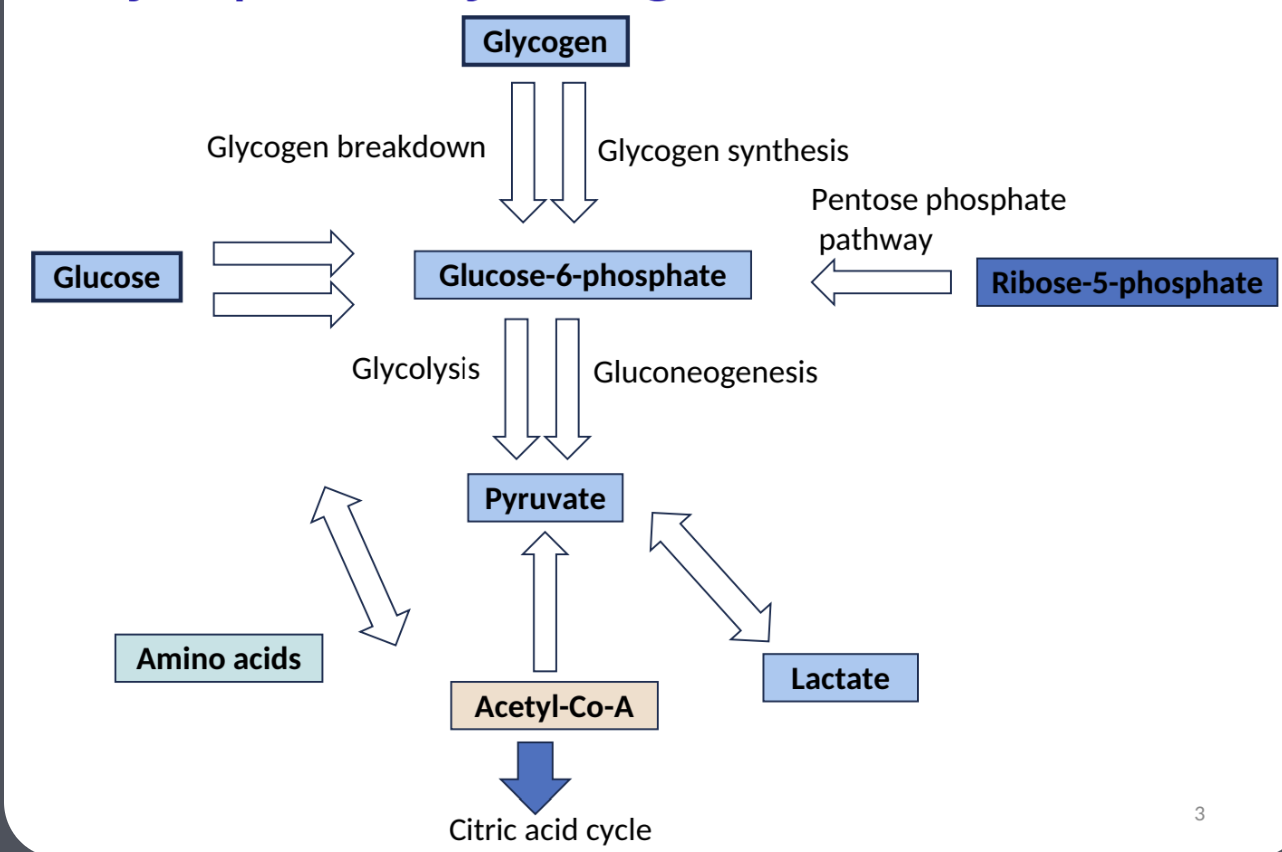
Major Pathways of Glucose Metabolism
What is Glycogen?
Structure of Glycogen
Glycogen is a multi-branched polysaccharide that serves as a form of energy storage in animals.
It consists of glucose units linked together.
Function of Glycogen Storage
Why Cells Store Glucose as a Polymer
Osmotic pressure is impacted by the concentration of glucose in a cell.
Glycogen can represent up to 10% of liver wet weight, which corresponds to a high concentration of free glucose.
Maintaining glucose in polymer form limits the osmotic pressure, preventing liver cell swelling and bursting.
By linking glucose units, the osmotic effect is diluted, allowing efficient storage without damaging cellular integrity.
Overview of Glycogen Metabolism
Key Enzymes and Pathways
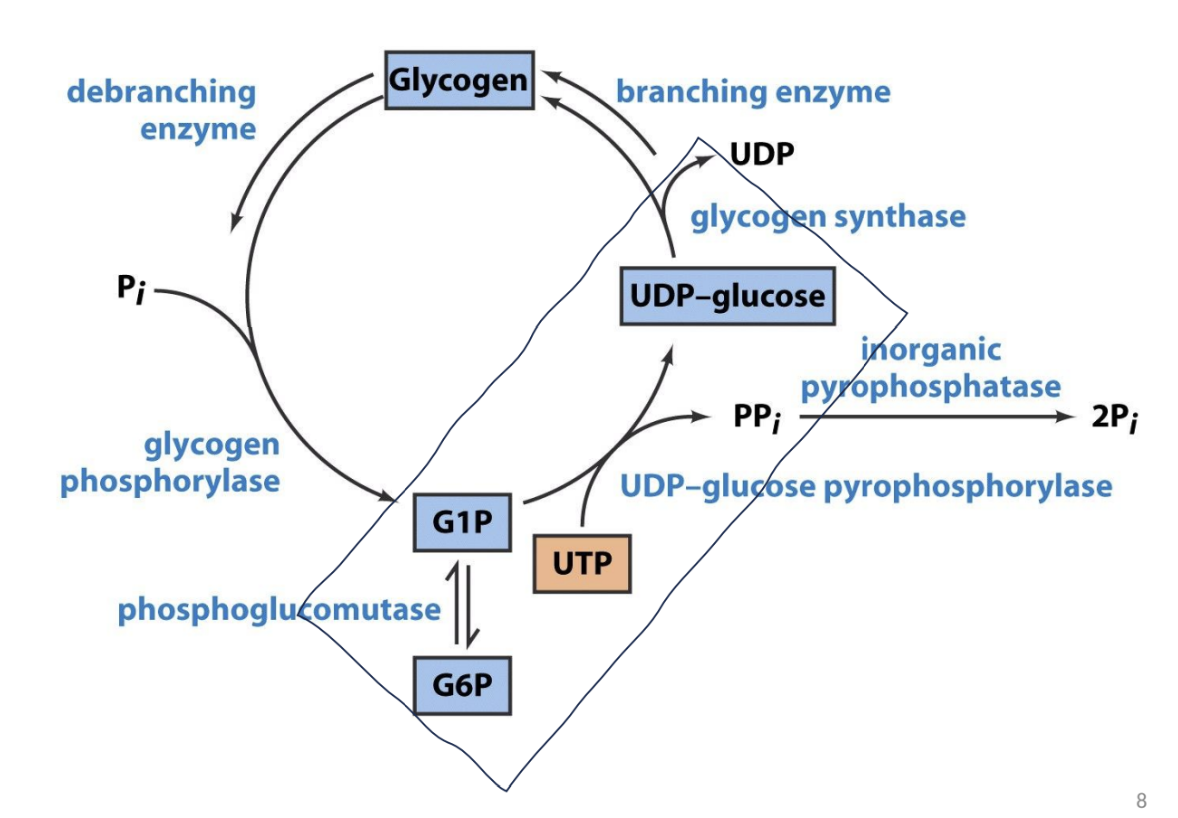
Main Enzymatic Players:
Glycogen branching enzyme
Glycogen synthase
Glycogen phosphorylase
Inorganic pyrophosphatase
Phosphoglucomutase
Glycogen Synthesis
Role of Glycogenin and Branching Enzymes
Glycogenin is essential for initiating glycogen synthesis.
UDP-glucose acts to elongate glycogen chains through glycogen synthase, promoting a branched structure through the action of branching enzymes.
Glycogen Degradation
Glycogen breakdown is facilitated by glycogen phosphorylase and involves debranching enzymes to fully mobilize glucose-1-phosphate for energy use.
Regulation of Glycogen Metabolism
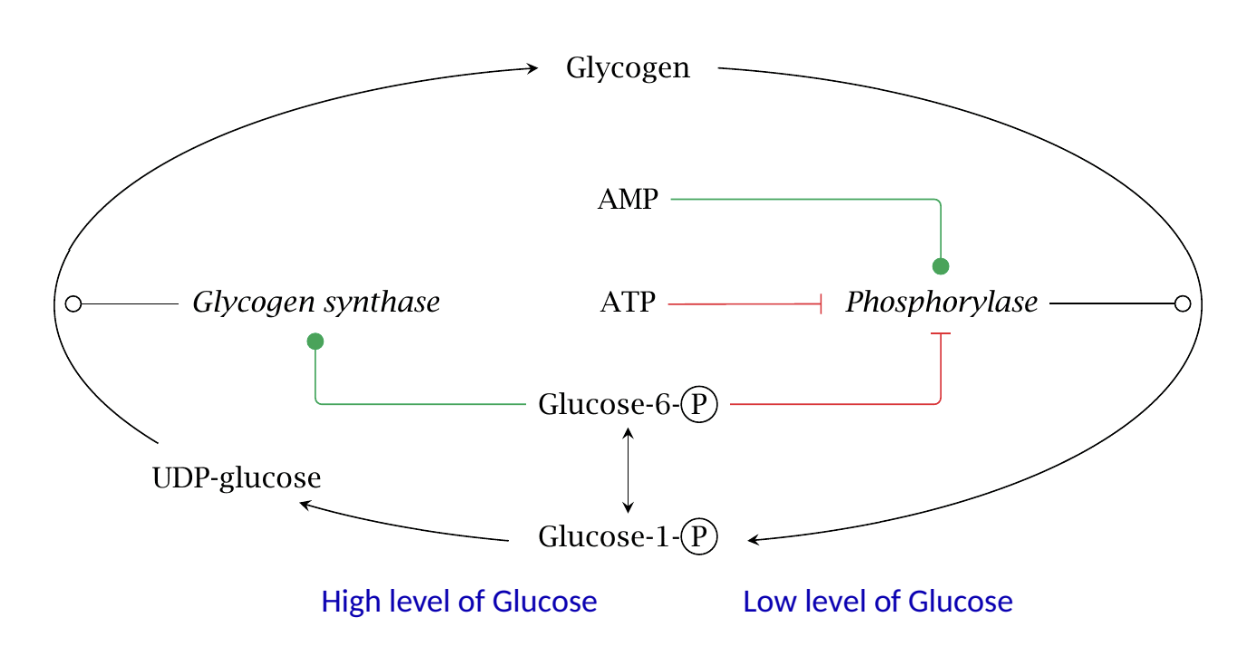
Allosteric Regulation
High levels of glucose promote glycogen synthase activity, while low glucose levels activate glycogen phosphorylase.
Hormonal Control
Key hormones influencing glycogen metabolism: Epinephrine, Glucagon, and Insulin.
These hormones trigger pathways that affect glycogen synthesis and breakdown, mediated through secondary messengers like cAMP.
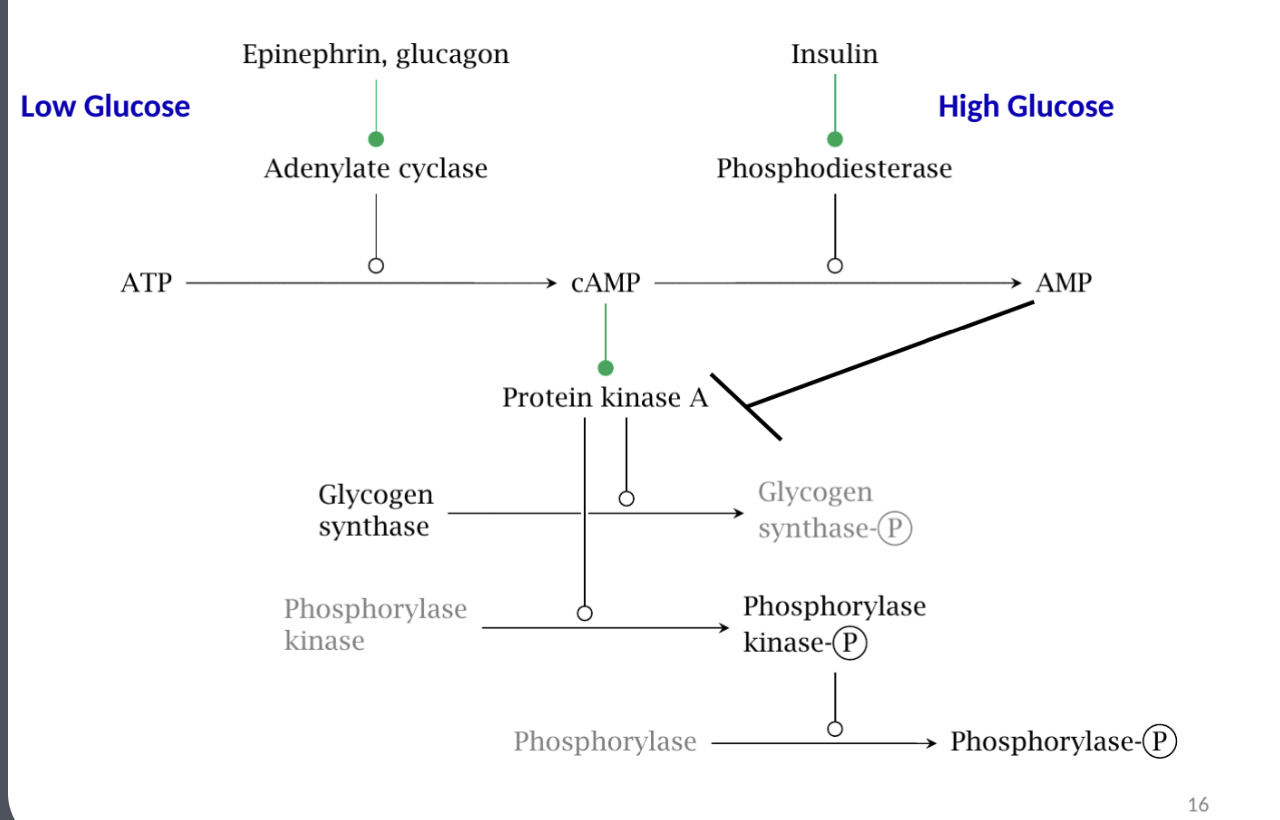
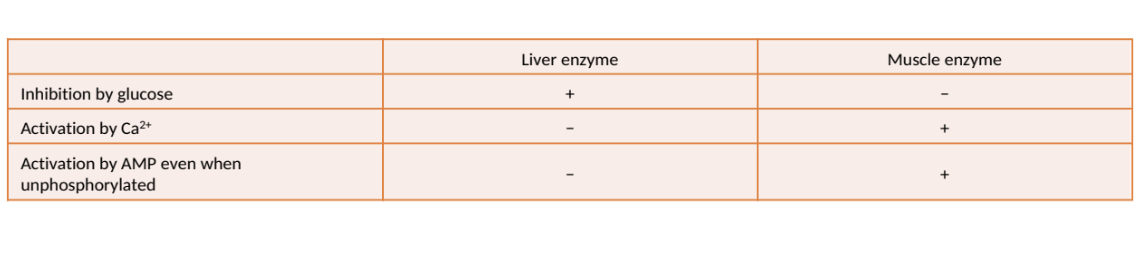
Phosphorylase Regulation in Liver & Muscle
Glycogen Storage Diseases
Overview
Glycogen storage diseases are inherited disorders due to defective enzymes involved in glycogen metabolism.
Enzyme deficiency results in abnormal glycogen accumulation, impairing liver and muscle function.
Important Types of Glycogen Storage Diseases
Type 0 (Glycogen Synthase Deficiency): Low blood glucose; early death.
Type Ia (von Gierke): Enlarged liver; kidney failure due to a lack of glucose-6-phosphatase.
Type II (Pompe): Muscle and heart-related issues due to deficiency of glucosidase.
Type III (Cori or Forbes): Debranching enzyme deficiency causing liver and muscle problems.
Type V (McArdle): Skeletal muscle phosphorylase deficiency; exercise-induced cramps and myoglobinuria.
Summary of Type Ia (von Gierke) Disease
Characterized by defect in glucose-6-phosphatase, leading to significant accumulation of glycogen.
Symptoms include profound liver enlargement, hypoglycemia, and metabolic derangements, including increased lactate levels.
Glucose Transport and Homeostasis
Glucose-6-phosphate's dephosphorylation in the ER allows effective transport and regulation of glucose homeostasis.
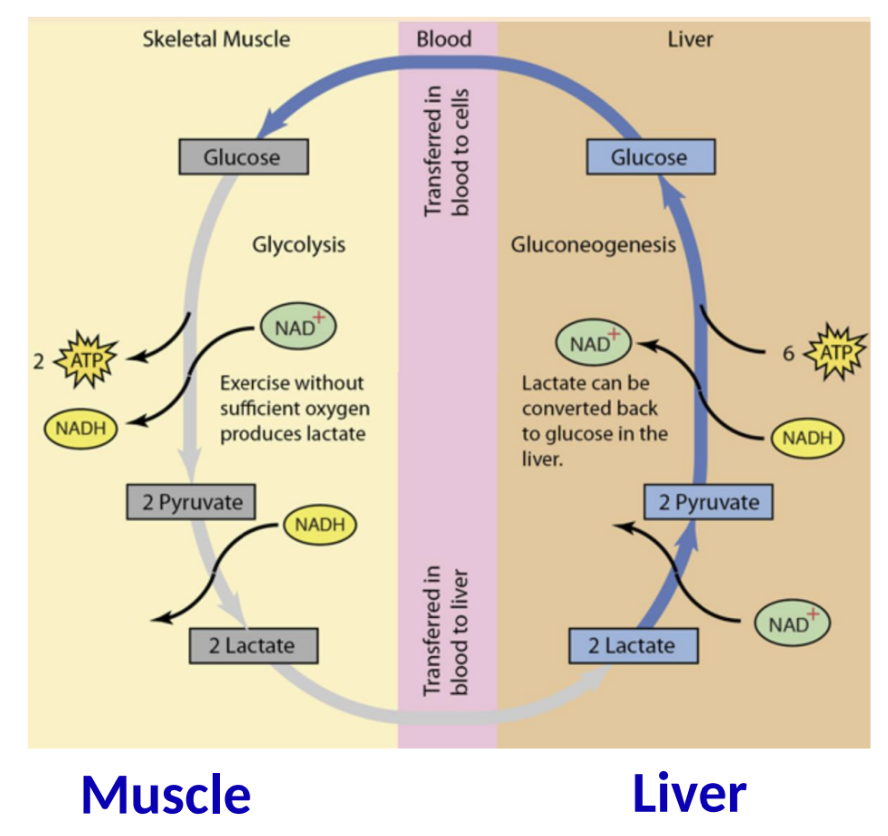
Muscle and Liver Collaboration
In exercise, skeletal muscle can convert lactate back to glucose in the liver, demonstrating the interconnectedness of metabolic pathways across tissues.
Learning Outcomes Recap
Reinforced understanding of glycogen structure, regulation, glycogen storage diseases, and their implications on metabolic health.