Lectures 22 and 23
Reaction Kinetics:
Outlines:
Principle of Detailed Balance: This principle helps us understand how reactions behave at a very fundamental level, especially when they can go in both forward and reverse directions. It's like saying that at equilibrium, every individual forward process is balanced by its exact reverse process. This is applicable when analyzing reversible reactions to relate kinetic and thermodynamic properties.
Rate Equations: These are mathematical descriptions that tell us how fast the concentrations of reactants and products change over time. They are crucial for predicting how a reaction will proceed and how quickly it will finish. Applicable for quantifying reaction speed and understanding factors affecting it.
Molecularity and Elementary Reactions: Molecularity describes how many molecules are involved in a single step of a reaction (e.g., unimolecular means one molecule, bimolecular means two). Elementary reactions are the basic, individual steps that make up a more complex overall chemical change. Understanding these helps us build up the overall mechanism. Applicable to define reaction steps and their kinetic order.
First Order Reactions: Reactions where the rate depends only on the concentration of one reactant, raised to the power of one. For instance, if Rate =k[A]=k[A], it's first order with respect to A. Many decay processes, like radioactive decay, follow first-order kinetics. Applicable for predicting reactant consumption in simple unimolecular decompositions.
Second Order Reactions: Reactions where the rate depends on the concentration of two reactants (each raised to the power of one, e.g., Rate =k[A][B]=k[A][B]) or one reactant raised to the power of two (e.g., Rate =k[A]2=k[A]2). Many common bimolecular reactions in solution follow second-order kinetics. Applicable for understanding how two molecules interacting affect reaction speed.
Important Dates:
Exam 2 Date: November 5 (Wednesday)
Location: Biochemistry Building Rm 101
Seating & Distribution: Starts at 10:10 AM
Exam Duration: 10:20 AM - 11:20 AM
Exam Coverage: Materials up to October 31
Review Session: November 3 (Monday)
Review materials available the night before.
Exam Structure:
Types of Questions:
Mixture of multiple choice, short answer, true/false questions
Two to three subquestions for each of the 7 big questions (+ bonus questions)
Provided Materials:
Basic equations, constants, hints during the exam
No outer resources allowed; calculator usage is optional.
Reaction Mechanisms
Definition of Mechanism: A reaction mechanism is like a step-by-step recipe or story that describes exactly how a chemical reaction happens at the molecular level. It tells us which bonds break, which bonds form, and in what order. Each step is an "elementary reaction." This is applicable for understanding the real, microscopic details of how molecules transform during a reaction.
Goal of Kinetics Study: The main goal of studying chemical kinetics (the study of reaction rates) is often to figure out this detailed mechanism of a chemical reaction, typically by trying to find and identify any temporary, unseen molecules called intermediates. Once we know the mechanism, we can control or optimize the reaction. This is applicable in designing efficient chemical processes or catalysts.
Example Reaction: Let's look at how hydrogen iodide (HI) might form from iodine (I2) and hydrogen (H2):
Overall Reaction: I2+H2→2HI
Conceptual Question: Why do we need a mechanism if we already have an overall reaction?Answer: The overall reaction just shows what you start with and what you end with, but not the path taken. The mechanism shows the actual molecular journey.
Overall Rate Constant: koverall (This is the rate constant for the entire reaction process, which is often a combination of the rate constants of the individual steps.)
Step 1 (Elementary Reaction): I2⇌2I
Explanation: First, an iodine molecule (I2) splits into two iodine atoms (I). These iodine atoms are highly reactive and are considered intermediates because they are formed and then quickly consumed in subsequent steps.
Conceptual Question: Why is this step reversible?
Answer: Bonds can break, but they can also reform. The two iodine atoms can collide and stick back together to form I2. So, this step can go both forward and backward.
Step 2 (Elementary Reaction): 2I+H2→2HI
Explanation: The two highly reactive iodine atoms (I) then collide with a hydrogen molecule (H2) to form two molecules of hydrogen iodide (HI).
Conceptual Question: What kind of reaction would this be based on molecularity?
Answer: It's a termolecular reaction in the way it's written, involving three particles (two I atoms and one H2 molecule). However, in practice, termolecular reactions (involving three molecules colliding simultaneously) are very rare. Mechanisms are often broken down into bimolecular (two molecules collide) or unimolecular (one molecule breaks apart) steps.
This step might actually happen in sub-steps (e.g. I+H2→HI, then H+I→HI), but for simplicity, it's shown as one step here.
Kinetic Constants:
Notations for Rate Constants: We use symbols like k1, k−1, k2 to represent the speed of each individual elementary step in a reaction mechanism.
k1: This usually refers to the rate constant for the forward direction of the first step.
k−1: This refers to the rate constant for the reverse direction of the first step (if it's reversible).
k2: This refers to the rate constant for the second step.
Conceptual Question: What does a large value of k1k1 tell us compared to a small value?
Answer: A large k1 means that step happens very quickly.
A small k1 means it's a slow step.
These constants are specific for each reaction step at a given temperature. They are applicable for quantifying the intrinsic speed of each elementary reaction, allowing us to compare the speeds of different steps within a mechanism.
Common Approximations for Kinetics of Intermediates:
When dealing with reaction mechanisms that involve intermediates, their concentrations can be very difficult to calculate directly because they are constantly being formed and consumed. So, we use approximations to simplify the math.
Steady-State Approximation (SSA):
Assumption: This approximation assumes that the concentration of the intermediate ([I]) remains constant throughout most of the reaction. It's not that the intermediate isn't reacting; it's that the rate at which it's being formed is equal to the rate at which it's being consumed.
Imagine filling a leaky bucket: if water flows in at the same rate it leaks out, the water level remains constant, even though water is constantly moving in and out.
Equation: Mathematically, this means the net rate of change of the intermediate's concentration is zero:
d[I]/dt=0Conceptual Question: When is it reasonable to make this assumption?
Answer: This is a good assumption when an intermediate is highly reactive and doesn't build up to high concentrations. It's formed, and then immediately reacts further, so its concentration stays consistently low over time.
Applicability: The SSA is widely used to derive overall rate laws for complex reactions when short-lived intermediates are involved. It simplifies systems of differential equations into algebraic equations.
Equilibrium Approximation (EQA):
Assumption: This approximation assumes that the formation of the intermediate is a reversible step that reaches equilibrium very quickly, much faster than the subsequent steps that consume the intermediate. This means the forward and reverse rates for that particular step are essentially equal.
Example Reaction: For our iodine dissociation step:
I2⇌2IEquilibrium Constant: At equilibrium, the ratio of products to reactants (each raised to their stoichiometric coefficients) is constant, and this constant is the equilibrium constant, Keq.
Expression: The equilibrium constant can also be expressed as the ratio of the forward and reverse rate constants for that step:
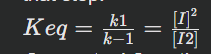
Conceptual Question: How is this different from the steady-state approximation?
Answer: SSA assumes the net rate of change of the intermediate is zero. EQA assumes a specific step (the one forming the intermediate) is at equilibrium, meaning its forward rate equals its reverse rate.
Applicability: The EQA is useful when an initial step in a mechanism is very fast and reversible, allowing us to express the intermediate's concentration in terms of reactants and the equilibrium constant. This simplifies the overall rate law.
Kinetic Analysis of Mechanisms
First Step: To analyze a general reaction mechanism, our main goal is to figure out the overall rate law for how a product is formed. The very first thing we do is write down a mathematical expression (a rate equation) for the concentration of any intermediate(s) involved in the reaction. This intermediate is a temporary molecule that is created and then used up during the reaction, so we need to understand how its amount changes.
From Reaction Steps: Let's look at our example reaction again:
I2⇌2I (forward rate constant k1, reverse rate constant k−1)
2I+H2→2HI (rate constant k2)
Now, let's write the rate of change for our intermediate, [I]. This means we want to know how fast the concentration of I is increasing or decreasing over time. We look at every step where I is created or consumed:
Rate of I formation from Step 1 (forward): +2k1[I2] (We get 2I atoms for every I2 molecule).
In the first step, one molecule of I2I2 breaks apart to form two I atoms. So, for every I2 molecule that reacts forward, we gain two I atoms. The rate constant k1 tells us how fast this splitting happens, and [I2] is the concentration of iodine molecules available to split.
Conceptual Question: Why is there a '2' in front of k1[I2]?
Answer: Because the product of this elementary step is 2I. For every mole of I2 that converts, two moles of I are produced.
Applicability: This term helps quantify how quickly the intermediate II is generated from the initial reactant I2I2.
Rate of I consumption from Step 1 (reverse): −2k−1[I]^2 (Two I atoms combine to form one I2)
Explanation: This is the reverse of the first step. Two I atoms combine to form one I2 molecule. So, I is being used up. The rate depends on [I]^2 because two I atoms need to collide for this to happen (bimolecular reaction of I with I). The rate constant k−1 tells us how fast this recombination occurs.
Conceptual Question: Why is this term negative and why is [I] squared?
Answer: It's negative because I is being consumed. It's squared because two I species are required to collide and react in this elementary reverse step.
Applicability: This term accounts for the loss of intermediate I when it converts back into the reactant I2.
Rate of I consumption from Step 2: −2k2[I]^2[H2] (Two I atoms react with one H2 molecule)
Explanation: In the second step, two I atoms react with one H2 molecule to form HI. Again, I is being consumed. The rate depends on [I]^2 (because two I's are needed) and [H2] (because one H2 is needed). The rate constant k2 governs the speed of this step.
Conceptual Question: How does the molecularity of Step 2 affect this rate term?
Answer: Step 2 is written as a termolecular reaction (2I+H2). This means the rate expression should be proportional to the concentrations of all three colliding species, hence [I]^2[H2]. However, real termolecular reactions are rare; often such steps are actually a sequence of bimolecular steps.
Applicability: This term describes how quickly the intermediate I is used up to form the final product HI.
Net Rate Equation: Combining all these contributions, the total rate of change for the intermediate [I] is:
d[I]/dt=2k1[I2]−2k−1[I]^2−2k2[I]^2[H2]Conceptual Question: Why is tracking the intermediate's concentration so important?
Answer: Intermediates are the transient species that form and are consumed during a reaction. Their concentrations and how they change over time provide direct evidence for the existence of specific steps in a proposed reaction mechanism. By understanding intermediates, we can identify the rate-determining step, which is the slowest step in the mechanism and controls the overall reaction rate. This information is vital for validating a mechanism, optimizing reaction conditions, and even designing catalysts to enhance reaction efficiency or selectivity by facilitating the formation or consumption of these critical intermediates.
Applicability: This full differential equation is the starting point for mathematically analyzing the reaction mechanism, especially when applying approximations like Steady-State or Equilibrium.
Desired Rate Equation for Product Formation: Once we have the rate equation for intermediates, our ultimate goal is to express the rate of formation of the final product (HI in our example). This desired rate law typically looks something like:
Rproduct = koverall[I2]^α[H2]^β
Explanation: Here, Rproduct is the rate at which the product (e.g., HI) is formed. koverall is the overall rate constant for the entire reaction, and [I2] and [H2] are the concentrations of the initial reactants. The exponents α and β (known as the reaction orders) tell us how sensitive the reaction rate is to changes in the concentrations of I2 and H2, respectively. These α and β values are determined experimentally or derived from the mechanism, and they are usually not the same as the stoichiometric coefficients in the overall reaction equation.
Applicability: This final rate law is what we would measure in an experiment. It allows us to predict reaction speeds under different conditions and provides a testable hypothesis to validate our proposed mechanism.
Steady-State Approximation
Concentration Activity of Intermediate Remains Constant: This approximation is incredibly useful when an intermediate is very reactive and short-lived. It assumes that the concentration of the intermediate ([I]) doesn't change significantly over most of the reaction's duration. This means that the rate at which it's being formed is approximately equal to the rate at which it's being consumed.
Set Up Equation: Mathematically, this assumption is expressed by setting the net rate of change of the intermediate's concentration to zero:
racd[I]/dt = 0
Conceptual Question: If d[I]/dt = 0, does it mean the intermediate isn't reacting at all?
Answer: No, it means the intermediate is reacting, but the rate of its formation is exactly balanced by the rate of its consumption. It's like a stable population in ecology where birth rate equals death rate.
Applicability: By making d[I]/dt = 0, we turn a complex differential equation into a simpler algebraic equation, which is much easier to solve.
Solving for Intermediate's Concentration: Applying SSA to our net rate equation for [I] from the previous section:
d[HI]/dt=2k2[I]^2[H2]
Now, substitute the expression for [I]^2 derived from SSA:
d[HI]/dt = 2k2[(k1[I2])/(k−1+k2[H2])][H2]
This simplifies to the overall rate law for product formation:
Rproduct=(2k1k2[I2]H2])/(k−1+k2[H2])
Conceptual Question: How does this final rate law compare to Rproduct=koverall[I2]α[H2]βRproduct=koverall[I2]α[H2]β? Answer: This specific derived rate law is the form we were aiming for, where koverallkoverall is actually the complex term rac2k1k2k−1+k2[H2]rac2k1k2k−1+k2[H2], and the reaction orders $\alpha and $\beta for [I2][I2] and [H2][H2] are 1, respectively. However, if k2[H2]k2[H2] is much smaller than k−1k−1, the denominator simplifies to k−1k−1, making it a more standard-looking rate law. If k2[H2]k2[H2] is much larger than k−1k−1, the denominator simplifies to k2[H2]k2[H2], and the rate law becomes rac2k1k2[I2][H2]k2[H2]=2k1[I2]rac2k1k2[I2][H2]k2[H2]=2k1[I2].
Applicability: This procedure allows us to predict how the overall rate of reaction depends on the concentrations of starting materials and the elementary rate constants, providing a testable model against experimental data.
Equilibrium Approximation
If the formation of an intermediate is reversible: This approximation is used when an initial step in a reaction mechanism is very fast and reaches equilibrium much faster than the subsequent steps that consume the intermediate. It essentially means that the forward and reverse rates of that specific step are equal to each other.
Utilize equilibrium constant for reaction: For our first step, I2⇌2I, if it quickly reaches equilibrium, we can define an equilibrium constant, Keq.
Keq=[products]/[reactants]=[I]2/[I2]
Explanation: At equilibrium, the ratio of the product concentrations to the reactant concentrations (each raised to their stoichiometric coefficients) is a constant value, Keq. In this case, I is the 'product' of this equilibrium step and I2 is the 'reactant'.
Conceptual Question: How is Keq fundamentally related to the rate constants?
Answer: At equilibrium, the forward rate equals the reverse rate. So for I2⇌2I, k1[I2]= k−1[I]^2.
Rearranging this gives:
k1/k−1=[I]2/[I2]
Thus, Keq=k1/k−1.
Applicability: The equilibrium constant provides a direct way to relate the intermediate's concentration to the reactant's concentration without dealing with differential equations for that specific step.
Solving for Intermediate's Concentration: From the equilibrium constant expression, we can easily solve for [I]2:
[I]^2=Keq[I2]
Conceptual Question: How is this different from the expression for [I]^2 we got from the Steady-State Approximation?
Answer: The EQA expression is simpler; it directly relates [I]^2 to [I2] and a single constant Keq. SSA typically results in a more complex expression involving multiple rate constants and concentrations, reflecting a dynamic balance rather than a static equilibrium for that step.
Applicability: This simpler expression for [I]^2 makes the subsequent substitution into the product rate law much more straightforward.
Rate Expression (Product Formation): Now, substitute this simpler expression for [I]² into the rate law for product formation (rate of HI production from Step 2):
d[HI]/dt = 2k2[I]²[H2]
Substituting [I]² = Keq[I2]:
d[HI]/dt = 2k2(Keq[I2])[H2]
Which gives us:
Rproduct=(2k2Keq)[I2][H2]
Explanation: In this case, the overall rate constant koverall becomes 2k2Keq. The derived rate law shows that the reaction is first order with respect to both [I2] and [H2].
Applicability: This method is powerful when an initial step is much faster than subsequent steps, simplifying the kinetic analysis and providing a direct link between the equilibrium of an initial step and the overall reaction rate.
Principle of Detailed Balance
What it means: At equilibrium, for a reversible reaction, the principle of detailed balance states that every single elementary step in the forward direction is balanced by its exact reverse step. It's not just that the overall rates of products forming and reactants disappearing are equal; it means that each individual back-and-forth mini-reaction is happening at the same speed.
Conceptual Question: Why is "each individual elementary step" important, and not just the overall reaction rates?
Answer: If the overall reaction rates were balanced but not the individual steps, it would imply that at equilibrium, some forward steps could be happening faster than their reverse, and other reverse steps faster than their forward, somehow "averaging out." This is physically unrealistic at a microscopic level. Detailed balance ensures that at equilibrium, the system is truly static on average, with no net flow in any specific microscopic pathway.
Applicability: This principle is crucial for rigorously connecting microscopic reaction mechanisms to macroscopic equilibrium properties. It's a fundamental concept in chemical kinetics and statistical mechanics.
Connecting Thermodynamics and Kinetics: This principle acts as a bridge between how fast reactions happen (kinetics) and where they settle over long periods (thermodynamics).
Gibbs Free Energy (ΔGrxn∘): This tells us about the energy change in a reaction under standard conditions and predicts whether a reaction is spontaneous (happens on its own) and how far it will proceed to products or reactants at equilibrium.
Equation: ΔG∘rxn=−RTlnKeq
Here, ΔGrxn∘ is the standard Gibbs Free Energy change for the reaction.
R is the ideal gas constant (8.314J/molK).
T is the temperature in Kelvin.
Keq (Equilibrium Constant) is a ratio of product concentrations to reactant concentrations at equilibrium, each raised to their stoichiometric coefficients. It quantifies the extent of a reaction at equilibrium.
Conceptual Question: What does a large value of Keq (e.g., Keq>1) tell us about the reaction at equilibrium, and how does this relate to ΔGrxn∘?
Answer: A large Keq means that at equilibrium, there are more products than reactants. This implies the reaction tends to proceed extensively in the forward direction. Mathematically, if Keq>1, then lnKeq is positive, making ΔG∘rxn negative. A negative ΔG∘rxn indicates a spontaneous reaction that favors product formation at equilibrium. Conversely, if Keq<1, there are more reactants, and ΔGrxn∘ is positive, meaning the reaction is non-spontaneous in the forward direction under standard conditions and favors reactants at equilibrium.
Applicability: This equation allows us to predict the spontaneity and equilibrium composition of a reaction from its thermodynamic properties, which is vital for process design.
Arrhenius Equation (krxn): This equation deals with kinetics, specifically how temperature affects the rate constant (krxn) of a reaction.
Equation: krxn=Ae^(−Ea/RT)
krxn is the rate constant, which tells us how fast a reaction proceeds at a given temperature.
A is the pre-exponential factor (or frequency factor), which reflects how often molecules collide with the correct orientation to react. Think of it as the maximum possible rate if there were no energy barrier.
Ea is the activation energy, representing the minimum energy required for reactants to transform into products. It's the "energy hurdle" they must overcome.
R and T are the gas constant and temperature, as before.
Conceptual Question: How do A and Ea influence the reaction rate constant, and why does temperature play such a big role?
Answer: A larger A means more frequent and correctly oriented collisions, leading to a faster reaction. A smaller Ea means a lower energy barrier, so more molecules have enough energy to react, also leading to a faster reaction. Temperature is critical because the exponential term −Ea/(RT) is very sensitive to T. As temperature increases, the kinetic energy of molecules increases, leading to more frequent and energetic collisions, and a larger fraction of molecules possessing enough energy to overcome Ea, thus significantly increasing krxn.
Applicability: This equation is fundamental for predicting reaction rates at different temperatures and for understanding the energy requirements for a chemical transformation.
Note on Rate Constants and Equilibrium: If Keq ≠ 1, it means the equilibrium favors either products or reactants. This also implies that the forward and reverse rate constants (k1 and k−1) are generally not equal, and therefore their corresponding Arrhenius parameters (A1, Ea1 for forward and A−1, Ea−1 for reverse) are also distinct.
Conceptual Question: If Keq ≠ 1, does it mean the forward reaction is intrinsically "faster" or "slower" than the reverse reaction?
Answer: Not necessarily "faster" or "slower" in an absolute sense, but rather that the energetic pathways are different. If Keq>1, it means the products are more stable than reactants (thermodynamically favored). This doesn't mean the forward activation energy (Ea1) is necessarily lower than the reverse (Ea,−1). What it does mean is that the ratio of the forward rate constant to the reverse rate constant (k1/k−1) is equal to Keq. Therefore, if Keq>1, then k1>k−1 at equilibrium. This implies the relative stability of reactants and products drives the equilibrium composition.
Applicability: This highlights that thermodynamics and kinetics are distinct but related. A thermodynamically favorable reaction does not guarantee a fast reaction, and vice versa.
Example for a Simple Reversible Reaction: A⇌B
Let's consider a simple reversible reaction where reactant A turns into product B, and B can turn back into A.Forward reaction:
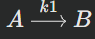
Where the rate constant is k1Reverse reaction:
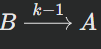
Where the rate constant is k-1
Rate Expressions: These differential equations describe how the concentrations of A and B change over time, considering both the forward and reverse processes.
Equation for A: d[A]/dt=−k1[A]+k−1[B]
Explanation:
−k1[A]: This term is negative because A is consumed by the forward reaction (A changing to B). The rate depends on the concentration of A.
+k−1[B]: This term is positive because A is formed by the reverse reaction (B changing back to A). The rate depends on the concentration of B.
Equation for B: d[B]/dt=k1[A]−(k−1[B])
Explanation:
+k1[A]: This term is positive because B is formed by the forward reaction (A changing to B).
−k−1[B]: This term is negative because B is consumed by the reverse reaction (B changing back to A).
Conceptual Question: Why are there two terms in each rate equation for a reversible reaction, and what do the signs indicate?
Answer: There are two terms because the concentration of each species (A and B) is affected by both the forward and reverse reactions. The signs indicate whether the species is being formed (positive term) or consumed (negative term) in that particular elementary step.
Applicability: These equations are the foundation for modeling and predicting the concentration changes of species in reversible processes over time.
At Equilibrium: According to the Principle of Detailed Balance, at equilibrium, the forward rate of each elementary step equals its reverse rate. For our simple example, this means:
Rate forward for A to B = Rate reverse for B to A
So, k1[A]eq = k−1[B]eq
The subscript "eq" denotes concentrations at equilibrium.
Conceptual Question: What does it mean for d[A]/dt (and d[B]/dt) to be zero at equilibrium?
Answer: When the system reaches equilibrium, the net change in the concentrations of A and B becomes zero. This means that A is being formed and consumed at exactly the same rate, and similarly for B. This state implies that there is no further macroscopic change in concentrations, even though the microscopic reactions (forward and reverse) are still happening actively.
Applicability: This direct relation at equilibrium allows us to define the equilibrium constant from intrinsic kinetic parameters: Keq=[B]eq/[A]eq = k1/k−1. This clearly shows the fundamental link between kinetics and thermodynamics inherent in the principle of detailed balance.
Kinetics of Reversible Reactions
Rate Equations:
Two differential equations for each species in reversible reactions:
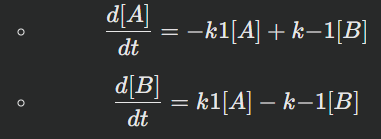
Rapid Dynamics of Reaction Species:
Time constants defined by rate constants determine behaviors of rising and falling exponentials for both reactants and products.
MATLAB Method for Solving Rate Equations
System Configuration:
With two rate equations for the species A & B, utilize symbolic algebra in MATLAB to analyze the changes in concentrations.
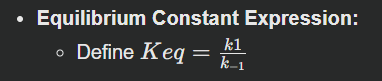
Extra Problems and Example Scenarios
