AP Chem Unit 3 (Thermodynamics)
Review Stations
Station #1
Vocabulary:
- Temperature: How hot something is; reflects the average transitional kinetic energy of molecules. Units: Kelvin or Celsius
- Heat: Thermal energy transfer. Does not necessarily mean hot. Units: Kilojoules or Joules
- Energy: Energy must be transferred for work to be done
- Endothermic: Absorbing heat or taking in energy. ∆H is +
- Exothermic: Releasing heat or releasing energy. ∆H is -
Heat transfer:
- Heat flows thermodynamically favorably from hot to cold places. Either by conduction or IR radiation
Enthalpy of Reaction:
- Endothermic: * ∆H+ * Reactants have less potential energy than the products * “heat” is a reactant * Bond breaking
- Exothermic: * ∆H- * Products have more potential energy than the reactants * “heat” is a product * Bond forming
Heating Curve:
- y-axis: temp
- x-axis: time
- Phase change: constant, ∆H fusion/∆H vaporization, potential energy increases, kinetic energy constant
- Temperature change: increase, q = mc∆t, potential energy and kinetic energy constant
E
Standard Enthalpies:
- ∆H Reaction (rxn): For molar quantities represented by the balanced equation given
- ∆H Combustion (comb): Per mole of substance burned in pure oxygen
- ∆H Vaporization/Fusion (vap/fus): Per mole of substance burned or melted
- ∆H Neutralization (neut): Per mole of water made during a neutralization reaction
- ∆H Solution/Heat of Solution (soln): Per mole of substance dissolved completely in water
- ∆H Formation/Heat of Formation (f): Per 1 mole of compound from its elements in standard state
\ \ \
Station #2
Specific Heat (c):
- The energy required to raise the T of one gram of a substance one degree Celsius * Specific heat of water: 4.18J/g˚C * Equation used for calculating the heat needed to change a substance by a given temp: q = mc∆T
- q sys = - q surr
Phase Changes:
- Equation used for calculation the heat needed to change a substances’s phase * Constants for water: * ∆H fusion: 6.01 kJ/mol * ∆H vaporization: 40.7kJ/mol
- Combined problems: * Draw a heating curve * Increase: phase stays same, temperature change (q = mc∆t); constant: phase change (∆H fusion or vaporization)
\
Station #3
Hess’ Law:
- The total energy change in a chemical reaction will be the same if it happens in one step or several steps.
- Toolbox: * A chemical reaction can be reversed * Sign for ∆H becomes opposite * A chemical reaction can be multiplied by a coefficient * ∆H also needs to be multiplied * A chemical reaction can be divided by a coefficient * ∆H also needs to be divided * Chemical reactions can be added together * If a chemical appears as both a reactant and product and are of the same value, they cancel out * After all that can be cancelled is cancelled out, the remaining products and reactants should match the given equation
Heats of Formation:
- “Enthalpy of Formation”
- ∆H of formation is the heat gained or lost when ONE compound is created from its elements under standard conditions (25˚C and 1 atm)
- \
\
Station #4
Thermochemical Equations
- Energy in Chemical Reactions
- Convert to moles of whatever is being produced/burned, then include the ∆H of the reaction into the equation
Bond Dissociation Energy (BDE)
- Energy is required to break the bonds in the reactants (endothermic) and released when forming bonds in products (exothermic)
- When ∆H is positive, that means you need more energy to break than to bond
- Calculate by adding up all the bond dissociation values
\
\
Station #5
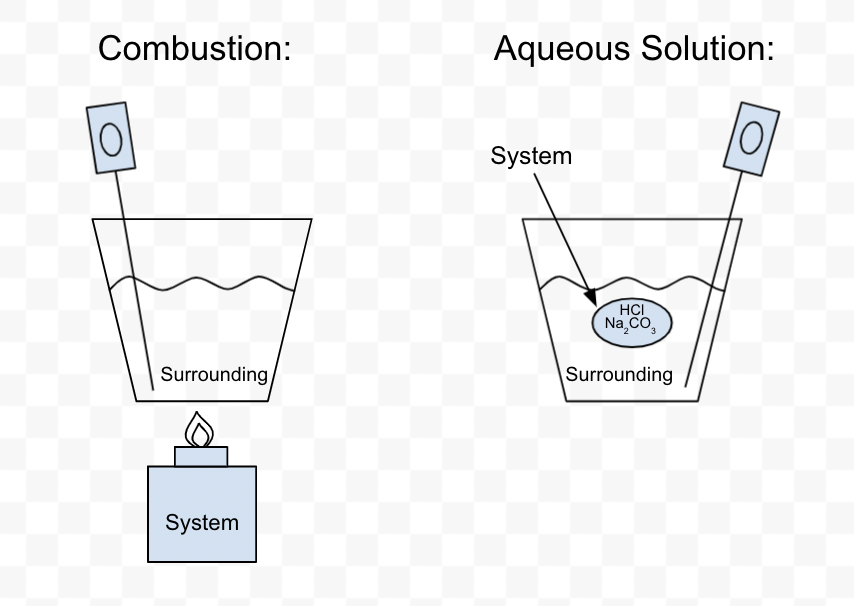
Measurement of Heat Flow (Calorimetry): System vs. Surroundings
- System: The change that we focus on
- Surroundings: All changes other than the system * Usually the water that surrounds the system
- Calorimeter: Instrument used to measure the amount of heat released or absorbed
- Heat Loss: Major source of error in all calorimetry experiments
- Typical Calorimetry Experiments
*
 Steps for calculating ∆H in typical calorimetry experiments
* Find q of Surroundings
* q = mc∆T
* Find q of the system
* q system = -q surroundings
* Find moles of the system
* ∆H = q system/moles system
Steps for calculating ∆H in typical calorimetry experiments
* Find q of Surroundings
* q = mc∆T
* Find q of the system
* q system = -q surroundings
* Find moles of the system
* ∆H = q system/moles system
\ \
\
\
Unit 3 Packet: Thermodynamics
Summation of Heat of Formation
∆H = ∑H products - ∑H reactants
\
Entropy
Entropy is the measure of the degree of disorder of a system or dispersal of matter or energy
- Phases: gas > liquid > solid
- \ # of moles: look at coefficients, more moles = more entropy
- Temperature: T increases, kinetic energy increases, entropy of system increases
- Molecular complexity: large molecules have more complexity and more entropy
Entropy changes:
- ∆S = +: increase in entropy/more entropy, corresponds to more disorder (less order)
- ∆S = -: decrease in entropy/less entropy, corresponds to less disorder (more order)
Summation of Entropy:
- ∆S = ∑S products - ∑S reactants
\
Second Law of Thermodynamics
- The entropy of an isolated system does not decrease.
- The entropy of the universe must increase
- ∆S universe = ∆S system + ∆S surroundings
- ∆S surroundings = ∆H/T
\ \
Third Law of Thermodynamics
- The entropy of a perfect crystal at absolute zero is exactly equal to zero
- Only a perfect crystal at 0˚K has no entropy
\
Spontaneity
A Spontaneous reaction proceeds to completion without any outside help
- ∆H (+), endothermic: non- spontaneous, needs energy to be put in for the reaction to happen
- ∆H (-), exothermic: spontaneous
- ∆S (+), more disorder: spontaneous, the isolated system must increase
- ∆S (-), less disorder: non-spontaneous
\
Gibbs’ Free Energy (∆G)
The max amount of useful work that can be obtained from a process at standard conditions
- ∆G (+) = non-spontaneous, not thermodynamically favorable, reaction won’t happen
- ∆G (-) = spontaneous, thermodynamically favorable, reaction will happen
Relationship Between Entropy, Enthalpy, and Gibbs’ Free Energy
- ∆G = ∆H - T∆S * T in K, ∆G and ∆S in kJ * \
| ∆H | ∆S | Spontaneous @… |
|---|---|---|
| - | - | lower temp |
| + | + | higher temp |
| + | - | never |
| - | + | always |
- Summation of ∆G * ∆G = ∑G products - ∑G reactants
\
Enthalpy/Entropy Driven
- ∆G = ∆H - T∆S
- If ∆H and ∆S favor opposite processes, spontaneity will depend on temperature
- Enthalpy driven: ∆H and ∆S are negative * Spontaneous at low temp, exothermicity is dominant * When ∆H is -, Enthalpy drives the reaction because a negative ∆H is thermodynamically favorable (releasing energy)
- Entropy driven: ∆H and ∆S are positive * Spontaneous at high temp, exothermicity is relatively unimportant * When ∆S is +, Entropy drives the reaction because a positive ∆S is thermodynamically favorable (more disorder)
- ∆S is what makes ∆G negative * When ∆G is negative, the reaction is favorable/spontaneous
\
Equilibrium and ∆G
At equilibrium ∆G = 0. This is the point where a reaction will become thermodynamically favorable or not favorable
- Set ∆H and T∆S equal to 0
- At low temp, ∆H drives the reaction (enthalpy)
- At high temp, ∆S drives the reaction (entropy)
\