Lecture 2.1 pt 1
Techniques for analyzing nucleic acids Absorption of UV light
Spectrophotometry (UV absorption)
Common technique for measuring DNA: spectrophotometry
DNA absorbs UV light strongly at 260 nm; most other macromolecules (proteins, carbs, lipids) do not.
Procedure: place sample in a spectrophotometer and set to 260 nm; measures light absorbed/transmitted.
Higher absorbance indicates more nucleic acid/DNA present (higher concentration).
Can also be used to assess purity of DNA:
Proteins and other contaminants absorb at 280 nm.
Purity is assessed by the ratio of absorbance at 260 nm to absorbance at 280 nm (A260/A280), where a ratio of approximately 1.8 is typically considered pure DNA.
Typical interpretation: a higher ratio indicates purer DNA; common reference values depend on sample type.
Example (absorbance values given): Which would you want?
Abs260 Abs280
Sample 1 0.75 1.5
Sample 2 1.5 1.5
Sample 3 0.5 0.1
Which would you want?
Sample 1: 0.75/1.5 = 0.50 → protein-rich; good protein sample, bad DNA/RNA.
Sample 2: 1.5/1.5 = 1.00 → mixed/contaminated; high nucleic acid amount but not pure.
Sample 3: 0.5/0.1 = 5.00 → unrealistic for nucleic acids; likely measurement/blank error or weird contaminant; also low concentration.
You want the heightest which is 5.00 however, in reality further dilution or purification may be necessary to obtain more accurate results (1.8-2.00)
Centrifugation
Centrifugation is a technique that uses a rapidly spinning rotor to generate a centrifugal force to move particles with mass toward/away from the rotor according to their mass and density.
Consequences: differential mass/shape/density cause separation of mixture components (DNA, protein, carbohydrate, lipid) during spinning.
Two major classes:
a) Sedimentation equilibrium centrifugation
b) Sedimentation velocity centrifugation
Analysis and purification
Sedimentation equilibrium centrifugation
Uses a density gradient (usually CsCl) created in the tube.
Mixture is spun for a fixed time; molecules separate based on buoyant density.
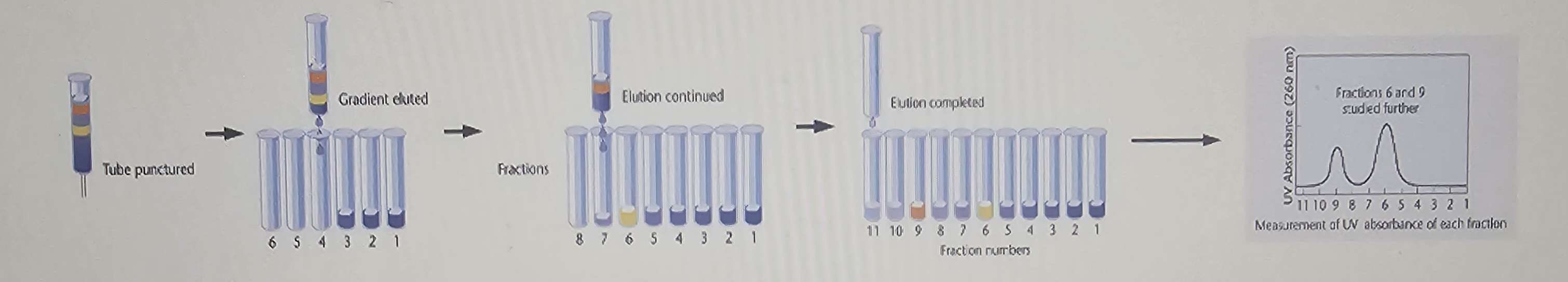
After spinning, fractions are collected by removing a hole at the bottom of the tube and measuring the presence of DNA or protein in each fraction.
Principle: separation depends on buoyant density (rho) of molecules in the gradient.
Sedimentation velocity centrifugation
Spinning creates a flow that moves molecules down the tube while being measured during centrifugation.
DNA/RNA shape and mass influence the rate of movement (sedimentation velocity).
DNA/RNA shape and mass both influence how fast it moves
Expressed in Svedberg units (S) –used to calculate mass
Denaturation and renaturation
Purpose: learn about DNA by separating strands and allowing them to re-assemble.
Denaturation: heat (or chemical treatment) disrupts hydrogen bonds between the two strands, separating them without breaking covalent (phosphodiester) bonds.
Renaturation: cooling allows strands to re-form hydrogen bonds and re-anneal to their complementary partners.
a) Determining relative ratios of A/T and GC content:
G and C form hydrogen bonds; A and T form hydrogen bonds.
Therefore, DNA with more G–C pairs denatures at higher temperatures due to stronger GC bonding.
It is not about the speed of denaturation but rather the bonds breaking due to the temp. as a whole.
b) Determining relative sequence similarity between two pieces of DNA:
Denature, mix, and allow renaturation; assess whether different DNA pieces hydrogen bond with each other (region of sequence similarity) vs. non-complementary regions (region of sequence difference).
Example question: Does gene X from humans have a similar sequence to the same gene in other species (e.g., apes)? Yes,it bc of the ability to link sequence data to evolutionary relationships
c) Determining sequence complexity:
Denature DNA and measure how long (or how quickly) it renatures.
More repetitive sequences renature faster because there are fewer unique binding partners.
This method helped identify repetitive DNA sequences (often referred to as “junk” DNA) that populate most human chromosomes between genes (e.g., AT-rich repeats like ATATAT…).
Garbage sequences: that do not code/translate for proteins and were historically considered non-functional but are now understood to play roles in regulation and chromosomal stability.
d) FISH (fluorescence in situ hybridization):
Design a probe that is specific to a DNA sequence (e.g., a gene).
Tag the probe with a fluorophore, florescent dye: that emits light at a specific wavelength, allowing for visualization of the probe's binding location under a fluorescence microscope.
Add to cells or chromosomes of interest; the probe will hybridize,anneal with its target sequence wherever it is located.
Add to cells (or chromosomes) of interest
Probe will (renature) with its target sequence wherever it is located
Location of fluorescence = Location of target
Major uses: determine if the target gene is present in the genome, find the gene’s location on a chromosome, and assess its expected location or abundance.
Radioactivity
Tag nucleic acids with various isotopes (radioactive labels) such as ; other options include radioactive isotopes of C, N, or H.
Tagging can be done in vivo or in vitro.
How each method is performed depends on the isotope and labeling strategy (e.g., incorporation into nucleic acids during synthesis).
Measurements are made with machines such as a scintillation counter to quantify radioactivity in samples.
Uses include tracking,measure,detect nucleic acids and studying biological processes; classic examples:
Hershey–Chase experiment
HSV-1 tracking
Concept: track the fate and localization of labeled nucleic acids or genomes in biological systems.
Gel electrophoresis
Method of separating molecules in a semi-solid gel under an applied electric field.
DNA fragments separate based on size/length because the gel matrix impedes movement proportionally to fragment length.
Procedure:
Load DNA fragments into a gel (usually agarose, a large polysaccharide derived from seaweed).
Apply an electric current; DNA (phosphates) is negatively charged and migrates toward the positive electrode.
The current forces the large DNA through to separate its fragments based on size, with smaller fragments moving faster through the pores of the gel than larger ones, allowing for the eventual analysis of the resulting bands.
Process:
Agarose (or polyacrylamide) gel has a network of pores.
Smaller DNA fragments move faster through these pores; larger fragments move slower.
Agarose gel forms a network through which DNA fragments migrate; smaller fragments navigate the matrix more easily and migrate faster than larger fragments.
DNA is negatively charged because of its phosphate backbone.
In gel electrophoresis, you apply an electric field with the negative electrode (cathode) at the top/well and positive electrode (anode) at the bottom.
DNA moves toward the positive electrode (opposite the negative electrode), not toward another negative current.
The combination of size (length of fragment) and charge determines how far each fragment moves.
Since all DNA fragments have roughly the same charge-to-mass ratio, size is the dominant factor in agarose gels.
Size:
Larger on top: Slower movement through the gel results in a higher position for larger fragments, while smaller fragments migrate faster and appear lower in the gel.
Smaller on bottom: As the DNA fragments move through the gel, the larger fragments experience more resistance, while the smaller fragments are able to migrate faster towards the bottom, creating distinct bands based on size.