intéractions protéine-protéine
les réactions protéines-protéines on des roles biologique importants:
Signal transduction
Cell growth and differentiation
Gene expression
Secretion
on a plusieurs méthodes pour étudier ces intéractions
méthodes d’étude d’interactions
Traditional Biochemical Methods:
Co-immunoprecipitation (Co-IP):
This technique is used to study protein-protein interactions by using an antibody against one protein to "pull down" that protein along with any proteins that it interacts with.
The interacting proteins are then identified through techniques like Western blotting or mass spectrometry.
Pull-down assays:
pour connaitre les interaction entre une protéine cible et les autres. La protéine testé doit être détectable. Quand on fait reproduire une protéine à travers un plasmide il faut lui mettre une étiquette — par exemple GST (Glutathione S-Transferase)
This method involves using a "bait" protein immobilized on a matrix (e.g., beads) to "pull down" interacting proteins from a complex mixture.
The bound proteins are then eluted and identified.
méthode du GST pull-down
voir à la fin
ce qu’on fait souvent c’est qu’on attache des étiquettes aux protéines, puis on met des anticorps anti l’étiquette pour qu’elles soient visiblent
Tandem Affinity Purification (TAP):
TAP is a two-step purification method used to isolate protein complexes.
The protein of interest is tagged with two affinity tags, allowing for stringent purification of the protein and its interacting partners.
Advanced Methods:
Yeast Two-Hybrid System (Y2H):
This is a powerful genetic method used to study protein-protein interactions in vivo (in yeast cells).
It relies on the reconstitution of a transcription factor when two proteins interact, leading to the expression of a reporter gene.
Colocalization and FRET (Fluorescence Resonance Energy Transfer):
These are microscopy-based techniques used to study protein interactions and their spatial organization within cells.
Colocalization involves visualizing two or more proteins labeled with different fluorescent dyes to see if they are present in the same location.
FRET is a technique used to measure the distance and orientation between two proteins by detecting the transfer of energy between two fluorescent dyes.
pull down
on va beaucoup l’employer, cf l’exemple
GST pull down
pour étudier les interactions protéine-protéine. Elle repose sur l'utilisation d'une protéine fusionnée à la Glutathion S-Transférase (GST), une enzyme qui se lie spécifiquement au glutathion.
idée: précipiter les protéines qui intéragissent, isoler les protéines et les identifier
Principe de la méthode
Préparation de la protéine fusionnée GST: Le gène codant pour la protéine d'intérêt est fusionné au gène codant pour la GST. La protéine fusionnée est ensuite exprimée dans un organisme hôte, généralement E. coli.
Préparation de l'extrait cellulaire: Les cellules sont lysées pour libérer les protéines, y compris la protéine fusionnée GST.
Incubation avec des billes de glutathion: L'extrait cellulaire est incubé avec des billes recouvertes de glutathion. La protéine fusionnée GST se lie aux billes, tandis que les autres protéines restent dans la solution.
Lavage: Les billes sont lavées pour éliminer les protéines non liées.
Élution: La protéine fusionnée GST est éluée des billes en utilisant une solution de glutathion libre.
Détection: La protéine éluée peut être détectée par différentes techniques, telles que l'électrophorèse sur gel et le western blot. Comme ça on dénature les protéines et elles se détachent des billes
Avantages de la méthode
Spécificité: La liaison entre la GST et le glutathion est très spécifique, ce qui réduit le risque d'interactions non spécifiques.
Facilité d'utilisation: La méthode est relativement simple à mettre en œuvre.
Polyvalence: La méthode peut être utilisée pour étudier une grande variété d'interactions protéine-protéine.
Inconvénients de la méthode
Interactions transitoires: La méthode peut ne pas détecter les interactions faibles ou transitoires.
Conditions in vitro: La méthode est réalisée in vitro, ce qui peut ne pas refléter les conditions physiologiques.
→ parfois on remplace ces bille de glutathion par des bille magnétiques et un aimant
limitations
Interactions non physiologiques :
Conditions in vitro : La méthode est réalisée in vitro, ce qui signifie qu'elle ne reproduit pas toujours fidèlement les conditions complexes et dynamiques de l'environnement cellulaire.
Absence de facteurs cellulaires : L'absence d'autres protéines et facteurs cellulaires peut influencer les interactions observées.
Artefacts : Des interactions non spécifiques peuvent se produire en raison de l'absence de compétition avec d'autres partenaires de liaison potentiels.
Sensibilité limitée à cause du lavage
Co-immunoprecipitation
utiliser un anticorps spécifique contre une protéine d'intérêt (appelée "appât") pour isoler cette protéine ainsi que toutes les autres protéines qui interagissent avec elle (appelées "proies")
Extrait de cellules entières : La Co-IP est réalisée en utilisant un extrait de cellules entières, ce qui signifie que toutes les protéines et autres composants cellulaires sont présents. Cela permet de maintenir les interactions protéiques dans un contexte physiologique, autant que possible.
Deuxième point : Une interaction positive reflète une véritable association in vivo.
Interaction positive : Si la Co-IP révèle que deux protéines sont présentes dans le même complexe, cela suggère qu'elles interagissent ensemble
mais attention une interaction positive ne signifie pas qu’elles sont en contacte
Déroulement de la Co-IP
Préparation de l'extrait cellulaire: Les cellules sont lysées pour libérer les protéines, y compris la protéine d'intérêt et ses partenaires d'interaction potentiels.
Ajout de l'anticorps: Un anticorps spécifique contre la protéine d'intérêt est ajouté à l'extrait cellulaire. Cet anticorps se lie à la protéine d'intérêt, formant un complexe immunitaire.
Capture du complexe immunitaire: Le complexe immunitaire est "capturé" à l'aide de billes recouvertes d'une protéine qui se lie à l'anticorps (par exemple, la protéine A ou G).
Lavages: Les billes sont lavées pour éliminer les protéines non liées, ne laissant que la protéine d'intérêt et ses partenaires d'interaction.
Élution: Les protéines liées aux billes sont détachées (éluées) en utilisant une solution qui perturbe l'interaction entre l'anticorps et la protéine d'intérêt.
Détection des protéines: Les protéines éluées sont ensuite analysées pour identifier les partenaires d'interaction de la protéine d'intérêt. Les techniques courantes incluent l'électrophorèse sur gel et le Western blot, qui permettent de séparer et d'identifier les protéines.
Applications de la Co-IP
Confirmer les interactions protéine-protéine suspectées : Si l'on pense que deux protéines interagissent, la Co-IP peut être utilisée pour le vérifier.
Identifier de nouvelles interactions protéine-protéine : La Co-IP peut révéler des interactions inattendues entre des protéines.
Étudier les complexes protéiques : Elle permet d'analyser la composition des complexes protéiques et d'identifier les protéines qui les constituent.
Comprendre les fonctions des protéines : En identifiant les partenaires d'interaction d'une protéine, on peut mieux comprendre sa fonction et son rôle dans les processus cellulaires.
Avantages de la Co-IP
Conditions physiologiques : La Co-IP est réalisée dans des conditions qui se rapprochent des conditions physiologiques, ce qui augmente la probabilité de détecter des interactions pertinentes.
Identification des partenaires d'interaction : Elle permet d'identifier et d'étudier les partenaires d'interaction d'une protéine spécifique.
Inconvénients de la Co-IP
Interactions non spécifiques : Des interactions non spécifiques peuvent se produire, conduisant à l'identification de protéines qui ne sont pas de véritables partenaires d'interaction.
Sensibilité limitée : La Co-IP peut ne pas détecter les interactions faibles ou transitoires.
Technique semi-quantitative : Elle ne donne pas d'informations quantitatives précises sur la force des interactions.
ensuite il faut analyser les protéines qu’on vient de tirer, souvent avec de la spétrométrie de masse
Tandem Affinity Purification" (TAP)
technique de marquage et de purification
La méthode TAP est une technique puissante pour étudier les interactions entre les protéines.
Elle utilise deux étiquettes d'affinité pour purifier efficacement les complexes protéiques, il y a donc deux étapes de purification
Elle permet une analyse à grande échelle des interactions protéiques, ce qui en fait un outil essentiel pour la protéomique. → pas à propos d’un gène mais de toutes les protéine d’un tissu
plus de détails
TAP : Tandem Affinity Purification (Purification par affinité en tandem)
Tandem :l'utilisation de deux étiquettes d'affinité (tags) distinctes, ce qui confère à la méthode une grande spécificité.
Les protéines sont marquées avec des étiquettes à haute affinité.
protéines modifiées génétiquement pour porter des étiquettes qui se lient à des substances spécifiques avec une très grande affinité.
Les constructions sont introduites dans les cellules hôtes.
Le terme "constructions" fait référence aux gènes modifiés qui codent pour les protéines fusionnées aux étiquettes TAP.
Les complexes protéiques sont purifiés sur des colonnes d'affinité.
Permet l'analyse à grande échelle des complexes protéiques.
La méthode TAP est particulièrement utile pour étudier un grand nombre de protéines et leurs interactions, ce qui en fait un outil précieux pour la protéomique, l'étude de l'ensemble des protéines d'un organisme.
tagging strategy
on utilise de cassettes
la cassette est un fragment d'ADN que l'on construit en laboratoire. Il contient plusieurs éléments importants :
Séquences loxP : Ce sont des marqueurs qui permettent de couper l'ADN à des endroits précis, grâce à une enzyme appelée recombinase Cre. On utilise ces séquences pour modifier l'ADN de manière ciblée.
Gène TRP1 : C'est un gène qui permet aux cellules de survivre dans un milieu de culture sans tryptophane (un acide aminé). On l'utilise pour sélectionner les cellules qui ont intégré la cassette dans leur génome.
Promoteur PGAL1 : C'est un interrupteur qui contrôle l'expression du gène situé juste après lui. Il est activé en présence de galactose, ce qui permet de produire la protéine désirée seulement quand on le souhaite.
Tags ProtA, TEV, CBP, EK : Ce sont des étiquettes qui se lient à des molécules spécifiques. On les utilise pour purifier la protéine qu'on veut étudier. Chaque tag a une affinité différente, ce qui permet de purifier la protéine de différentes manières.
Pourquoi utiliser une cassette ?
Modularité : La cassette est comme un assemblage de différents éléments, chacun ayant une fonction spécifique. On peut combiner ces éléments pour réaliser différentes expériences.
Contrôle de l'expression : Le promoteur PGAL1 permet de contrôler quand la protéine est produite. Cela peut être utile pour étudier les effets de la protéine à différents moments.
Facilité de manipulation : La cassette est plus facile à manipuler que de grands morceaux d'ADN. On peut l'amplifier, la modifier et l'insérer dans des cellules plus facilement.
étapes clé du tagging
PCR et transformation : La cassette est amplifiée par PCR, puis introduite dans des cellules (probablement de la levure, vu le marqueur TRP1).
Intégration du gène cible : La cassette s'intègre en amont du gène cible, sous le contrôle du promoteur PGAL1.
Expression de la protéine taguée : En présence de galactose, le promoteur PGAL1 est activé, entraînant l'expression du gène cible fusionné aux tags.
Purification : Les différents tags (ProtA, TEV, CBP, EK) permettent de purifier la protéine cible par affinité.
Recombinaison Cre : L'utilisation de la recombinase Cre permet de retirer une partie de la cassette, en laissant seulement le tag ProtA.
"Rapproche promoteur de région codante" : Après recombinaison, le promoteur est plus proche du gène cible, ce qui peut influencer son expression.
limitations
Le tag pourrait affecter la fonction de la protéine.
Le tag pourrait affecter le niveau d'expression de la protéine.
La protéine cible pourrait être clivée par la protéase TEV (enzyme utilisée pendant la purification)
The yeast two-hybrid system
principe simple : si deux protéines interagissent, on peut le détecter en utilisant un gène rapporteur.
Le système de double hybride de levure permet d'étudier les interactions entre deux protéines. On utilise des protéines chimériques qui rapprochent un domaine de liaison à l'ADN et un domaine d'activation transcriptionnelle si les protéines d'intérêt interagissent. L'interaction est détectée grâce à l'activation d'un gène rapporteur.
comment ça marche
formation de protéines chimérique (hybrides créées en combinant des domaines provenant de différentes protéines.)
Deux protéines "appâts" et "proies" : On a deux protéines que l'on veut étudier : une protéine "appât" et une protéine "proie". On fusionne chacune de ces protéines à un domaine protéique spécifique :
Protéine appât : Fusionnée au domaine de liaison à l'ADN d'un facteur de transcription.
Protéine proie : Fusionnée au domaine d'activation transcriptionnelle du même facteur de transcription.
Levure modifiée : On utilise une levure qui a été modifiée pour contenir un gène rapporteur (souvent un gène codant pour une enzyme) sous le contrôle du facteur de transcription.
Interaction et activation : Si la protéine appât et la protéine proie interagissent, elles rapprochent les deux domaines du facteur de transcription. Cela permet au facteur de transcription de se reformer et d'activer le gène rapporteur.
Détection : L'activation du gène rapporteur entraîne la production d'une enzyme ou d'une protéine détectable, ce qui indique que les deux protéines interagissent.
avantages
Relativement simple et peu coûteux
Analyse à grande échelle
Étude in vivo
Limitations
interactions artificielles → faux positif
Conditions non physiologiques
application
vérifier les interactions déterminées par des méthodes traditionnelles.
Cartographie fonctionnelle - domaines / acides aminés.
identifier les régions spécifiques des protéines qui sont impliquées dans l'interaction
Identifier de nouvelles protéines interagissantes (à partir d'une bibliothèque d'ADNc).
systèmes utilisés
nucléaire
GAL4
LexA
ce sont des facteurs de transcriptions avec domaine de liaison et domaine d’activation
membranaire:
SOS recruitment system (SRS)
pour voir les intérations entre protéines membranaires
basé sur un facteur d'échange GDP-GTP chez les mammifères. Il joue un rôle crucial dans l'activation de la protéine Ras, une protéine de signalisation impliquée dans la croissance cellulaire et d'autres processus.
Comment ça marche ?
Le SRS utilise ce principe pour étudier les interactions protéiques. Voici comment :
Protéine d'intérêt : Une des protéines dont on veut étudier l'interaction est fusionnée à hSos.
Protéine partenaire : L'autre protéine est ancrée à la membrane plasmique.
Interaction : Si les deux protéines interagissent, la protéine d'intérêt (fusionnée à hSos) est recrutée à la membrane plasmique, où se trouve son partenaire.
Activation de Ras : hSos, maintenant à la membrane, peut activer Ras.
Signalisation cellulaire : L'activation de Ras déclenche une cascade de signalisation qui aboutit à l'expression de certains gènes. On utilise souvent un gène rapporteur, comme le gène de la B-galactosidase, pour visualiser cette activation.
application
Interactions involving transcriptional activators or repressors
Interactions between proteins that require
modifications by cytoplasmic or membrane-
associated enzymes
faux résultat
le faux positif 1 (fausse activation)
Dans le cas présent, la protéine FISH, qui est censée se lier à l'ADN, est capable d'activer le gène rapporteur même en l'absence de la protéine "proie" fonction activation. Cela peut se produire si la protéine FISH possède une activité d'activation transcriptionnelle intrinsèque, ou si elle interagit avec d'autres protéines présentes dans la levure qui peuvent activer le gène rapporteur.
Après avoir observé une interaction positive, il est important de vérifier que la protéine FISH est capable d'activer le gène rapporteur seule, en l'absence de la protéine "appât". Si c'est le cas, cela signifie que le résultat est un faux positif.
le faux positif 2 (fausse liaison)
Dans le cas présent, la protéine FISH, qui est censée se lier à l'ADN, est capable d'activer le gène rapporteur même en l'absence de la protéine "appât" fonction de liaison. Cela peut se produire si la protéine FISH interagit avec d'autres protéines présentes dans la levure qui peuvent activer le gène rapporteur. Ici, la diapositive précise que la protéine FISH interagit avec le domaine de liaison de GAL4, une autre protéine de levure.
Après avoir observé une interaction positive, il est important de vérifier que la protéine FISH est capable d'activer le gène rapporteur seule, en l'absence de la protéine "appât". Si c'est le cas, cela signifie que le résultat est un faux positif.
le faux positif 3
Dans le cas présent, la protéine BAIT, qui est censée se lier à l'ADN, est capable d'activer le gène rapporteur même en l'absence de la protéine "proie". Cela peut se produire si la protéine BAIT interagit avec d'autres protéines présentes dans la levure qui peuvent activer le gène rapporteur. Ici, la diapositive précise que la protéine BAIT interagit avec le domaine d'activation de GAL4, une autre protéine de levure.
Avant de commencer le criblage, il est important de vérifier que la protéine BAIT est capable d'activer le gène rapporteur seule, en l'absence de la protéine "proie". Si c'est le cas, cela signifie que le résultat est un faux positif.
colocalisation
déterminer si deux protéines ou plus se trouvent au même endroit dans une cellule.
Immunostaining of two or more proteins on a fixed specimen
utilise des anticorps pour détecter des protéines spécifiques dans un échantillon fixé → traité
Look for overlapping staining
limitations: ce n’est pas parce que les protéines sont au même endroit qu’elles intéragissent forcément
FRET —fluorescence resonance energy transfer
mécanisme par lequel de l'énergie est transférée d'une molécule fluorescente (donneur) à une autre molécule fluorescente (accepteur) lorsque les deux molécules sont très proches l'une de l'autre.
Proteins are tagged with 2 different fluorophores. Une molécule fluorescente par protéines qu’on soupsconne qu’elles intéragissent.
→ on peut voir les intéractions dans des cellules vivantes
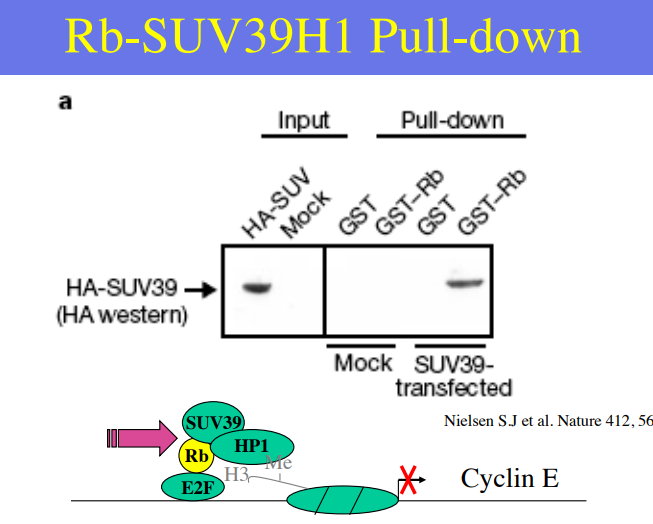
exemple (retinoblastoma)
c’est un cancer humain rare, qui commence pendant l’enfance. Loss of Rb (retinoblastoma) gene activity or Rb est un suppresseur de tumeur, elle doit absolument fonctionner. elle régule le cycle cellulaire
hyperphosphorylated in proliferating cells
hypophosphorlated (active) in differentiating cells
Pull-down : C'est une technique biochimique utilisée pour identifier les protéines qui se lient directement à une protéine d'intérêt
Dans ce cas, le schéma montre que les protéines SUV39, HP1 et E2F sont "tirées" avec Rb → suggère une intéraction directe ou indirecte

interpretation résultat
HA et GST sont des étiquettes
une étiquette HA est accrochée à la protéine. HA-SUV39 est la liaison entre l’étiquette et l’anticorps. Dans la première case on vérifie que ça marche avec l’anticorps et qu’on obtient rien sinon.
Dans la deuxième case on a un résultat que dans la dernière colonne, c’est un GST-Rb (protéine étiquettée) qui s’est liée à SUV39 parce qu’elles étaient présente. Dans Mock on a rien parce que les cellules ne produisaient pas de SUV39.
fabrication de protéines
on utilise des bactérie et des vecteurs pour produire des protéines
Clonage en phase :
Lorsque vous clonez un gène dans un vecteur pour l'expression de protéines, il est essentiel que le gène soit inséré dans le cadre de lecture correct par rapport au codon de démarrage du vecteur.
Cela garantit que le ribosome lira correctement les codons du gène et produira la protéine souhaitée.
→ s’il manque 1 ou 2 nucléotides c’est grave parce que ça déplace tout le carde de lecture.
spéctrométrie de masse
Elle repose sur la mesure du rapport masse/charge (m/z) des ions en phase gazeuse.
Principes de base de la spectrométrie de masse
Ionisation : Les molécules de l'échantillon sont ionisées, c'est-à-dire qu'elles gagnent ou perdent des électrons, ce qui leur confère une charge électrique. Il existe différentes techniques d'ionisation, telles que l'ionisation électronique (EI), l'ionisation chimique (CI) et l'électrospray (ESI). Le choix de la technique dépend du type de molécules à analyser.
Séparation : Les ions formés sont ensuite séparés en fonction de leur rapport m/z. Différents types d'analyseurs de masse peuvent être utilisés, tels que les analyseurs à secteur magnétique, les quadripôles, les pièges ioniques et les spectromètres temps de vol (TOF). Chaque type d'analyseur a ses propres avantages et inconvénients en termes de résolution, de sensibilité et de gamme de masses.
Détection : Les ions séparés sont détectés par un détecteur, qui mesure leur abondance. Le détecteur produit un signal électrique proportionnel au nombre d'ions détectés.
Analyse des données : Les données obtenues sont représentées sous forme d'un spectre de masse, qui est un graphique montrant l'abondance des ions en fonction de leur rapport m/z. L'analyse du spectre de masse permet d'identifier les molécules présentes dans l'échantillon et de déterminer leur quantité.
Library screening
Imaginez une immense bibliothèque contenant des milliers de livres. Vous cherchez un livre en particulier, mais vous ne connaissez ni le titre ni l'auteur. Le criblage de bibliothèque est comme une méthode pour trouver ce livre spécifique sans avoir à tous les lire un par un.
Les étapes clés
Construction de la bibliothèque
Introduction dans un système
Sélection ou criblage pour identifier les molécules qui possèdent la caractéristique ou la fonction recherchée
Amplification et identification : Les molécules sélectionnées sont amplifiées (par exemple, par PCR) et identifiées (par exemple, par séquençage)
types de criblage de bibliothèque
Criblage de protéines : Utilisé pour identifier des anticorps, des enzymes ou d'autres protéines qui se lient à une molécule spécifique.
Criblage d'ADN : Utilisé pour identifier des gènes, des promoteurs ou d'autres séquences d'ADN qui ont une fonction spécifique.
Criblage d'ARN : Utilisé pour étudier l'expression des gènes ou identifier des ARN non codants.