L1
Why Study Kinetics?
Thermodynamics only tells us whether a reaction is spontaneous; kinetics tells us how fast it proceeds. This distinction is critical because many spontaneous reactions occur at negligible rates without intervention.
Examples:
is thermodynamically spontaneous yet kinetically negligible over a human lifetime due to a very high activation energy.
Combustion of gasoline: extremely fast (useful for engines) due to a relatively low activation energy and high temperature.
Rust formation: very slow (affects materials over years) due to kinetic limitations and environmental factors.
Industrial relevance:
Faster processes ⟹ higher productivity & profit (e.g., flow chemistry for drug manufacture, optimizing industrial catalysts).
Slowing undesirable processes (food spoilage, corrosion, pollution) is equally important for product longevity and environmental protection.
Scientific & everyday relevance:
Drug-delivery systems: tailor release rates via nanoparticle design to ensure therapeutic concentrations are maintained over time.
Radiocarbon dating: kinetics of decay gauges fossil age, relying on its fixed half-life.
Fundamental Definitions
Chemical kinetics = study of reaction rates, mechanisms (pathways), and methods of control. It explores the molecular events that occur during a reaction.
Rate = change in a measurable quantity per unit time. This measurable quantity is typically the concentration (mol · L) of reactants or products, but can also be pressure (for gases) or absorbance (for colored solutions).
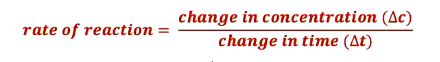
Generic everyday example: train speed (km · h).
Chemical example: change in concentration (mol · L) per second.
Reaction-Coordinate Diagram & Activation Energy
Reactants → Transition State(s) → Products. The transition state is a high-energy, unstable molecular arrangement that exists at the peak of the energy barrier. It is fleeting and cannot be isolated.
Activation energy = energy gap between reactants and transition state (height of the "hill"). It represents the minimum energy required for a reaction to occur.
Low EaEa
Only molecules with kinetic energy equal to or greater than E_a can successfully react upon collision.
Rate of Reaction: Formal Expression
For a single species :
Convention for a disappearance term (reactant):
Instantaneous rate: slope of the tangent to [A] vs. t curve at a specific time. This rate changes as the reaction proceeds and reactant concentrations change.
Average rate: slope of the straight line connecting two finite time points. This provides a general rate over an interval but doesn't capture instantaneous changes.
Stoichiometry & Relative Rates
For a general reaction
relative rates are linked by stoichiometric factors. The rate of the reaction is defined such that it is independent of which reactant or product is monitored:
Worked Stoichiometry Problems
Haber–Bosch:

Butane Combustion:

General Rate Law
Empirical relationship between rate and reactant concentrations (products generally do not appear in the rate law for elementary steps, though they can for complex mechanisms):
= rate constant: a proportionality constant specific for a given reaction at a particular temperature. Its value changes with temperature, often following the Arrhenius equation.
= reaction orders: exponents that indicate how the rate of reaction depends on the concentration of each reactant. These values must be determined experimentally and are not necessarily equal to the stoichiometric coefficients in the balanced chemical equation, especially for multi-step reactions. They reflect the elementary steps involved in the reaction mechanism.
Overall order =
0th, 1st, and 2nd orders are most common and the focus of this course.
Key Properties of Reaction Orders
Order | Exponent value | Rate dependence | Typical k units |
|---|---|---|---|
Zero | Rate independent of [A] | \text{mol·L}^{-1}\text{s}^{-1} (or M·s) | |
First | Rate [A] | ||
Second | Rate [A] or [A][B] | \text{L·mol}^{-1}\text{s}^{-1} (or M·s) |
Example – Using a Given Rate Law
Reaction: Pd-ligand complex binds a drug.
Empirically determined first-order law: with at 25 °C.
For :
\text{Rate}=0.09\times0.018=1.6\times10^{-3}\,\text{mol·L}^{-1}\text{h}^{-1}.
Instantaneous vs. Average Rate – Practical Tips
Draw the full concentration–time curve (or use data table) to visualize the rate changes.
Instantaneous: slope of tangent (graphical or derivative function) at a specific point in time.
Average: use two explicit time points in to calculate the overall rate over that interval.
Always include the minus sign for reactants so the final numerical rate is positive, as rates are defined as positive quantities.
Sign & Fraction Conventions Recap
Reactant terms carry a leading minus sign to ensure the overall rate is positive, as reactant concentrations decrease over time.
Each term is divided by its stoichiometric coefficient (convert coefficient to denominator fraction) to make the overall reaction rate independent of which species is being monitored.
Rates are always reported as positive numbers; signs appear only inside formal definitions to account for consumption or production.
Activation Energy & Pathway Control
Catalysis, temperature changes, solvent choice, and physical state all alter and hence rate. These factors are crucial for manipulating reaction speed:
Catalysis: A catalyst provides an alternative reaction pathway with a lower activation energy ( ). It does not change the overall thermodynamics of the reaction but significantly speeds up its rate.
Temperature: Increasing temperature increases the average kinetic energy of reactant molecules, leading to more frequent and more energetic collisions. A higher fraction of collisions will possess the required for reaction.
Surface Area: For heterogeneous reactions, increasing the surface area of solid reactants or catalysts provides more sites for molecules to interact, thereby increasing the rate.
Nature of Reactants: The inherent chemical properties and bond strengths of reactants influence the intrinsic .
Industrial goal: place reactions in the economic “sweet spot” (fast but controllable, minimal side-products, and efficient energy usage).
Societal challenges: slow down spoilage, oxidation, pollutant formation; speed up green manufacturing processes and waste detoxification.
Real-World Connections & Ethical Implications
Drug delivery nanoparticles need predictable release kinetics for safety and efficacy, ensuring drugs are released at a consistent and therapeutic rate.
Food security: kinetic inhibition (refrigeration, antioxidants, packaging) reduces waste by slowing down spoilage reactions.
Carbon-14 decay kinetics underpin archaeology and climate science; highlights interdisciplinary overlap in applying chemical principles.
Pollution control technologies (e.g., catalytic converters in cars) hinge on kinetic acceleration of detox pathways to convert harmful pollutants into less toxic substances more rapidly.