DP IB Chemistry: HL - Measuring Enthalpy Change
Difference Between Heat & Temperature
Heat vs. Temperature:
Temperature: Measure of the average kinetic energy of particles.
Heat: Measure of the energy content of a substance.
Example: Boiling water in a beaker vs. a drop; same temperature but different heat content due to quantity.
Kinetic Energy:
Particles have kinetic energy due to movement.
Faster movement means more energy and higher temperature.
Conservation of Energy:
Energy is the ability to do work.
Heat is one type of energy.
During chemical reactions, energy flows in and out of reaction vessels.
System and Surroundings:
System: Inside the reaction vessel.
Surroundings: Outside the reaction vessel.
Types of Systems:
Open: Matter and energy can move in and out (most common for chemical reactions).
Closed: Energy can move in and out, but matter cannot.
Isolated: Neither matter nor energy can be exchanged (very rare).
Law of Conservation of Energy:
Energy cannot be created or destroyed, only transferred.
Total energy remains the same, moving between the system and surroundings.
Exothermic & Endothermic Reactions
Enthalpy (Heat Content):
Total chemical energy inside a substance.
Represented by . Change in enthalpy is represented by .
Enthalpy Change:
Can be positive or negative.
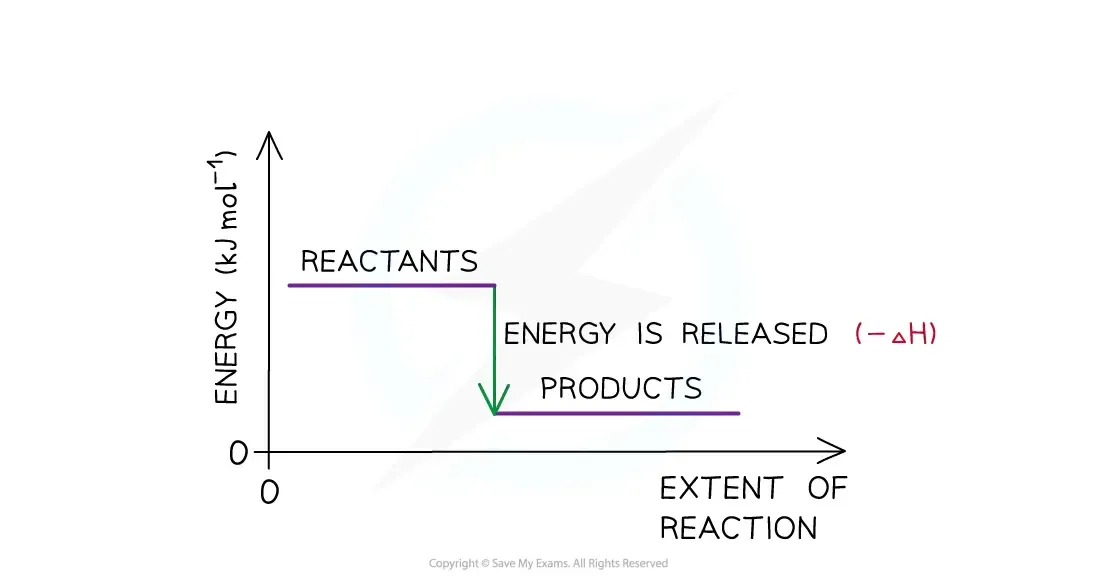
Exothermic Reactions:
Products have less enthalpy than reactants.
Heat energy is given off by the system to the surroundings.
Temperature of surroundings increases; temperature of the system decreases.
is negative.
Thermodynamically possible but may be kinetically controlled (rate too slow).
Energy flows out of the system.
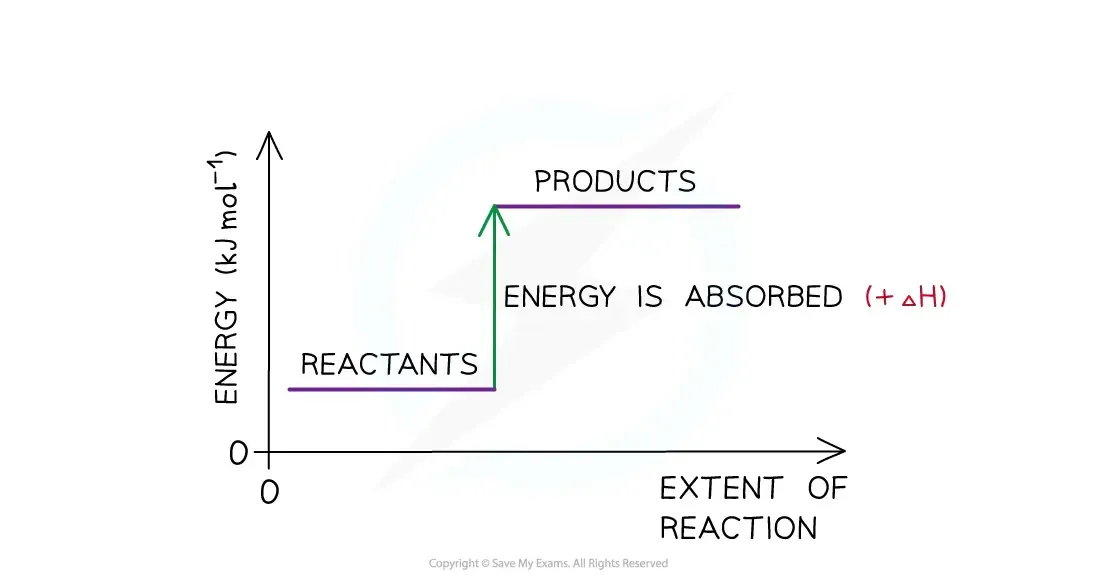
Endothermic Reactions:
Products have more enthalpy than reactants.
Heat energy is absorbed by the system from the surroundings.
Temperature of surroundings decreases; temperature of the system increases.
is positive.
System takes in energy, surroundings feel colder.
Key Reminder:
System = Reacting substances (the reaction itself).
Surroundings = Everything else (e.g., the flask).
Energy Profiles
Energy Profile Definition:
Shows energy changes in a chemical system during a reaction.
Includes energy of reactants, products, and transition state.
Transition State:
Stage where chemical bonds are partially broken and formed.
Unstable and cannot be isolated.
Highest energy point on the reaction pathway.
Activation Energy ():
Minimum energy needed to reach the transition state.
Defined as the minimum energy for reactant molecules to have a successful collision and start a reaction.
Exothermic Reaction Energy Profile:
Reactants are higher in energy than products.
Lower activation energy compared to endothermic reactions.
Products have less energy, releasing energy to surroundings.
Surrounding temperature increases.
Endothermic Reaction Energy Profile:
Reactants are lower in energy than products.
Higher activation energy compared to exothermic reactions.
Products have more energy, absorbing energy from surroundings.
Surrounding temperature decreases.
Worked Example 1: Enthalpy of Combustion for Methane
Methane combustion:
Enthalpy of combustion:
Activation energy:
Combustion is exothermic ( is negative), so reactants are drawn higher in energy than products.
Energy level diagram showing transition state.
Worked Example 2: Activation Energy Identification
Activation energy () is the energy difference from reactants to the transition state.
Enthalpy change of reaction is the energy difference from reactants to products.
Standard Enthalpy Change
Standard Conditions:
Pressure of 100 kPa.
Concentration of 1 mol dm$^{-3}$ for all solutions.
Each substance in its standard state (solid, gas, or liquid).
Temperature is usually given as 298.15 K.
Symbol is used to show standard conditions (e.g., ).
Standard Enthalpy Changes:
Formation (): Enthalpy change when one mole of a compound is formed from its elements under standard conditions (can be exothermic or endothermic).
Combustion (): Enthalpy change when one mole of a substance is burnt in excess oxygen under standard conditions (exothermic).
Neutralization (): Enthalpy change when one mole of water is formed by reacting an acid and alkali under standard conditions (exothermic).
Reaction (): The enthalpy change when the reactants in the stoichiometric equation react to give the products under standard conditions (can be exothermic or endothermic).
Worked Example 1:
Formation of one mole of water: ,
For two moles of water:
Worked Example 2:
Formation of 2 moles of Fe$2$O$3$:
Given
Worked Example 3: Identifying Enthalpy Changes
:
: and
:
Calorimetry Experiments
Calorimetry:
Technique to measure enthalpy changes of chemical reactions.
Calorimeter examples: polystyrene drinking cup, vacuum flask, or metal can.
Specific Heat Capacity ():
Energy needed to raise the temperature of 1 g of a substance by 1 K.
Specific heat capacity of water: 4.18 J g$^{-1}$ K$^{-1}$.
Heat Transfer Calculation:
= heat transferred (J).
= mass of water (g).
= specific heat capacity (J g$^{-1}$ K$^{-1}$).
= temperature change (K).
Types of Calorimetry Experiments:
Enthalpy changes of reactions in solution.
Enthalpy changes of combustion.
Enthalpy Changes for Reactions in Solution:
Carry out reaction with an excess of one reagent.
Measure the temperature change over a few minutes.
Worked Example: Propan-1-ol Combustion
0.01 mol of propan-1-ol heats 250 g of water from 298 K to 310 K.
Specific heat capacity of water = 4.18 J g$^{-1}$ K$^{-1}$.
Calculate values: m = 250g, c = 4.18 J g$^{-1}$ K$^{-1}$ , = 12 K
Energy released per mole:
Convert to kJ mol$^{-1}$:
Assumptions in Calculations:
Specific heat capacity of solution ≈ specific heat capacity of pure water (4.18 J g$^{-1}$ K$^{-1}$).
Density of solution ≈ density of pure water (1 g cm$^{-3}$).
Specific heat capacity of the container is ignored.
Reaction is complete.
Negligible heat losses.
Temperature Correction Graphs:
Used for non-instantaneous reactions to account for heat loss during the delay before maximum temperature is reached.
Extrapolate the cooling section of the graph back to the time when the reaction started to estimate the true maximum temperature.
Take temperature readings before adding reactants.
Add the second reactant and continue recording the temperature and time.
Plot graph and extrapolate cooling part to the time when the second reactant was added.
Assumption: Rate of cooling is constant.
Can also be used for endothermic reactions with a ‘warming’ section.
Enthalpy of Combustion Experiments:
Heat released by combustion reaction increases the heat content of water.
Use a simple calorimeter to measure temperature changes in water.
Worked Example:
Excess iron powder added to 100.0 cm$^3$ of 0.200 mol dm$^{-3}$ copper(II) sulfate solution.
Reaction:
Maximum temperature rise: 7.5 °C.
Calculate q:
Calculate moles of CuSO$_4$:
Calculate :
Sources of Error:
Heat losses to surroundings.
Heat absorbed by the calorimeter.
Incomplete combustion.
Minimizing Errors:
Do not place the copper calorimeter too far above the flame.
Use a lid over the calorimeter.
Use shielding to reduce draughts.
Worked Example: Propan-1-ol Combustion in Calorimeter
1.023 g of propan-1-ol (M = 60.11 g mol$^{-1}$) burned to heat 200 g of water; temperature rose by 30°C.
Calculate q:
Calculate moles of propan-1-ol:
Calculate :