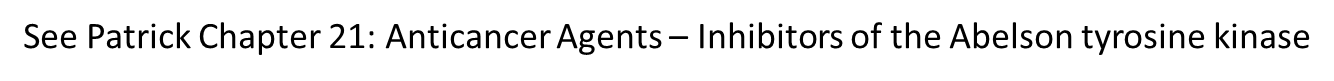Kinases (bcr-abl and imatinib)
8: Many responses not related to cancer involve an accelerated proliferative response eg arthritis
9: Kinase inhibitors currently in clinical use. bcr-abl = fusion protein targeted by imatinib (development started in early 90s, came into market in 2001)
one of big challenges with cancer is resistance developing to treatment due to change in protein structure over time (change in chemical space). This is one of reasons why nilotinib was developed as a later generation of imatinib. Many of inhibitors shown are 3rd or even 4th generation kinase inhibitors
The focus of kinase inhibitors is currently in cancer therapy but there are also rapidly emerging applications in inflammatory disease
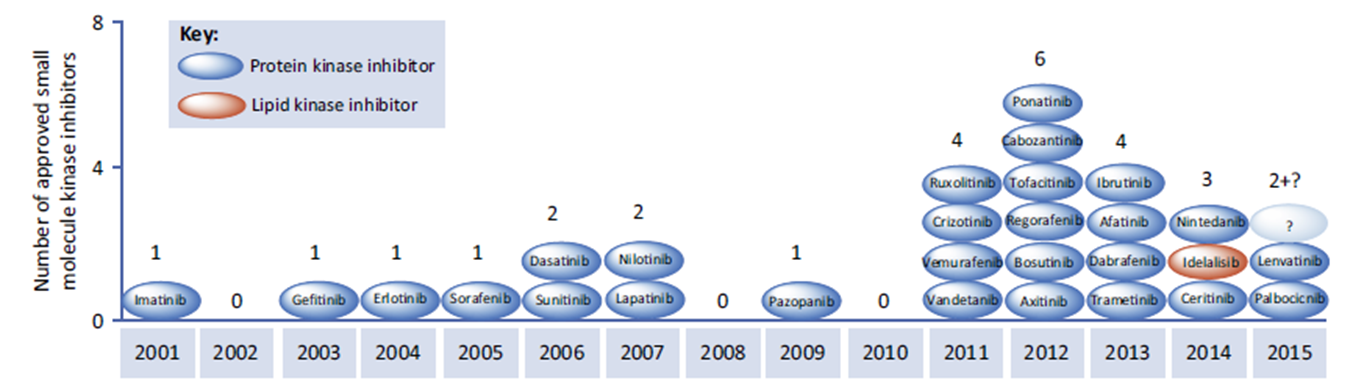
10: Kinase pathways
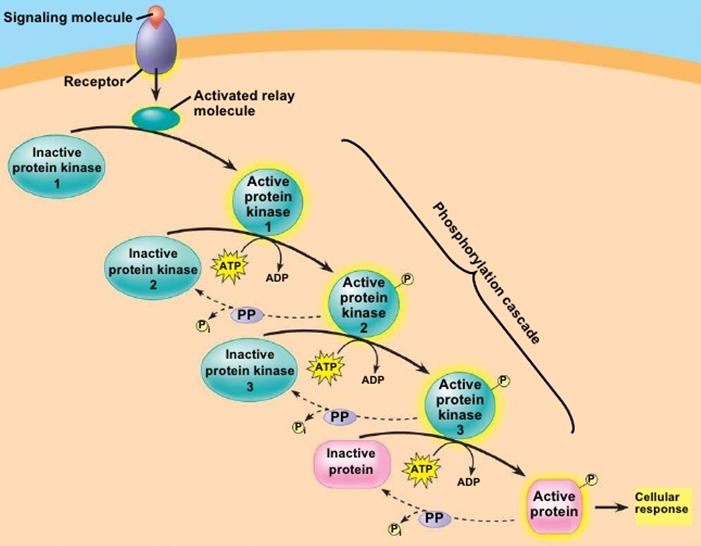
12: One of the consequences of phosphorylation by kinases is enzyme activation through causing a conformational rearrangement from active to inactive form.
They (kinases?) can activate themselves, typically after dimerisation when ligand bound eg EGF binding EGFR → dimerisation and autophosphorylation, converting EGFR into an active conformation. Activated EGFR can attract proteins to it via the phosphate group and phosphorylate other substrates to initiate signalling
13: Another consequence of kinase action is protein movement. Phosphate attachment can cause a protein to be attracted to other proteins which have positively charged motifs. They can be recruited to the membrane or nucleus to transport the signal around the cell.
If signalling goes wrong eg in cancer it can lead to survival of cancer cells despite/against host natural defence mechanisms, or abnormal proliferation and growth.
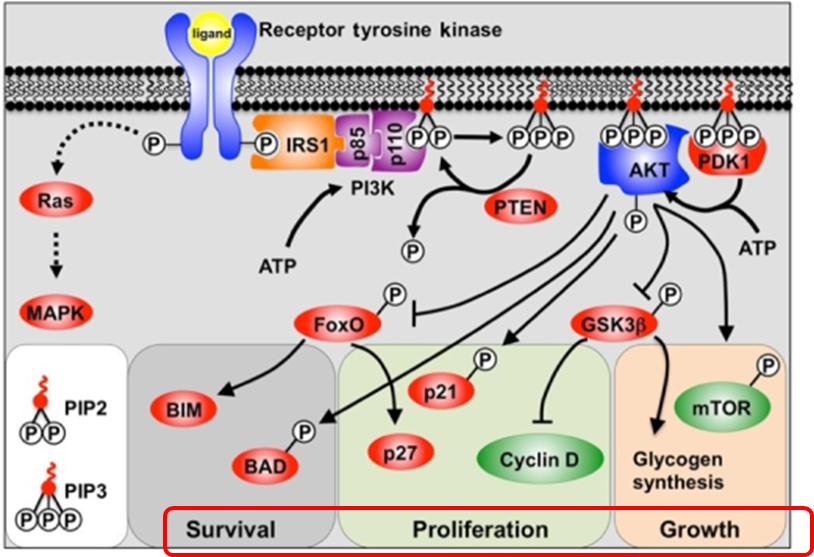
14: Turning signals off typically involves coordination between kinases and phosphatases. Typically kinases turn on and phosphatases turn off but this is pathway dependent and can be the opposite way around.
A particular kinase may have a critical role in a signalling pathway which promotes tumour cell abnormal growth/proliferation/survival. Stopping this kinase from phosphorylating its substrates could switch that signalling pathway off.
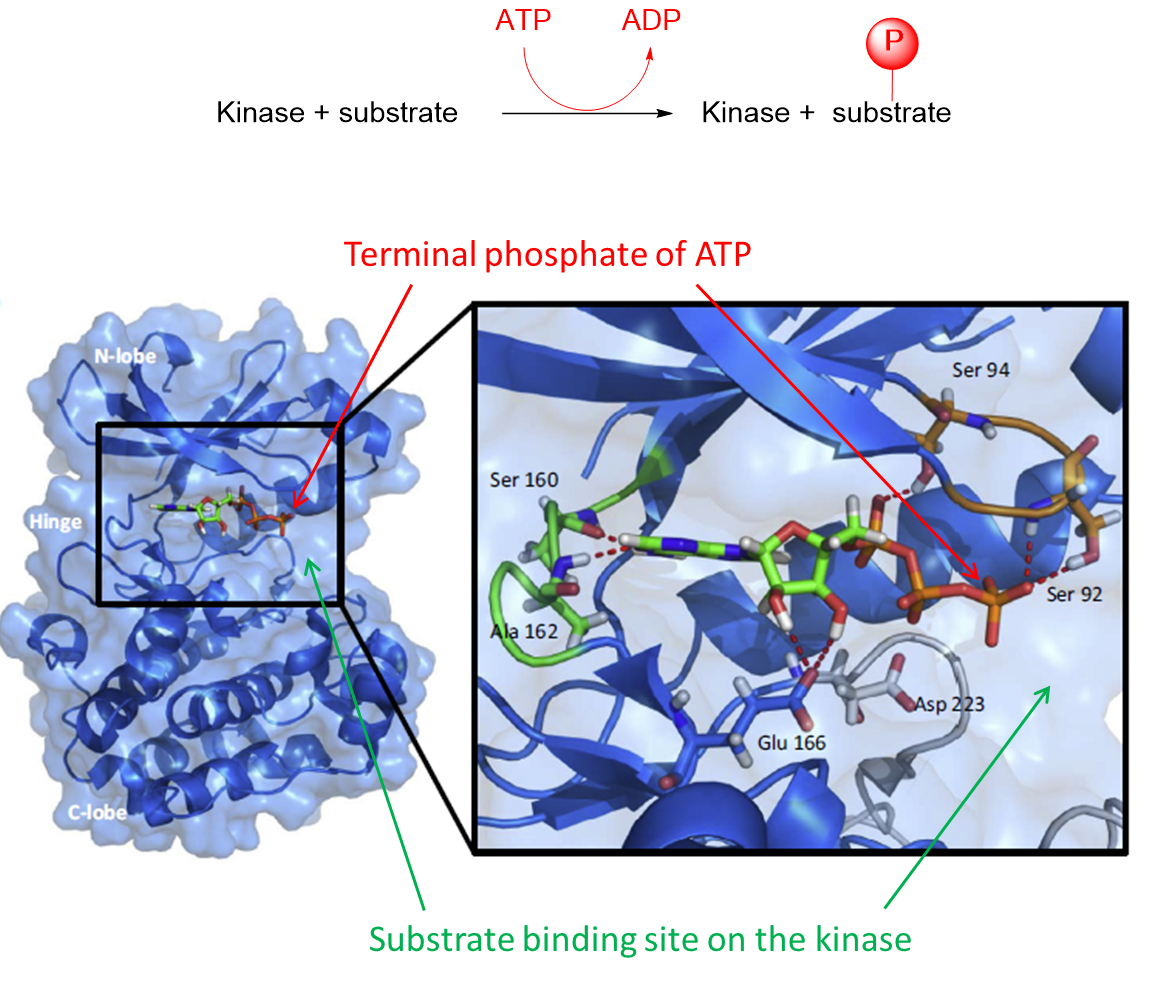
15: To stop a kinase from phosphorylating its substrate, the mechanism through which it enzymatically transfers phosphate must be understood. A molecule must be designed which prevents it from transferring phosphate and it must be made sure that only the kinase responsible for the dodgy signal is blocked in order to avoid off-target toxic effects.
16: All kinases have an overall bilobal global structure connected by a flexible hinge-linker. They all bind ATP as a cofactor but differ in specificity for their protein substrates which they phosphorylate.
ATP is bound at the hinge between the two lobes. The hinge closes around ATP and opens to release ADP - is this due to change in ATP conformation?
The activation loop binds protein substrate and positions it next to ATP so phosphate can be transferred.
Substrate binding site differs in every kinase allowing them to selectively bind and phosphorylate their specific associated substrate
17:
18:
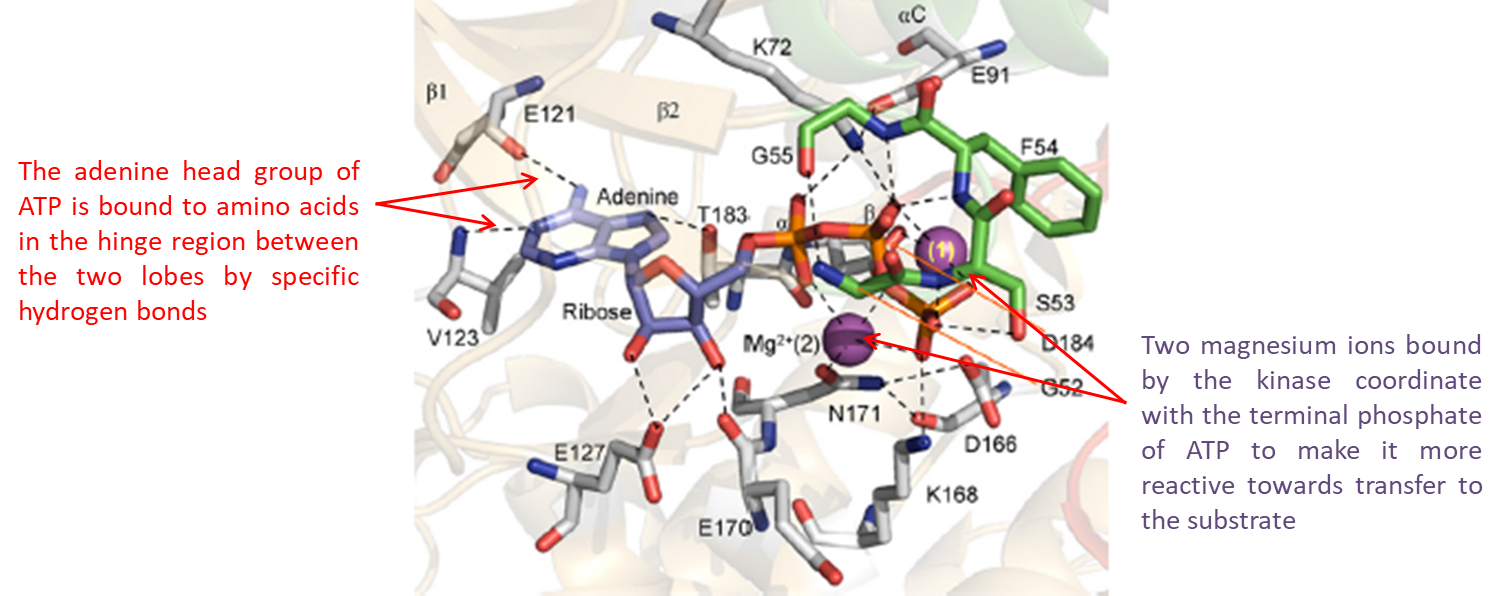
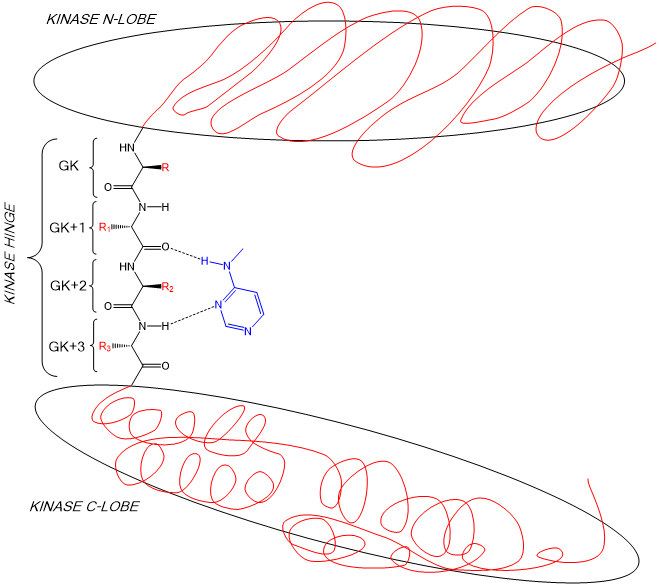
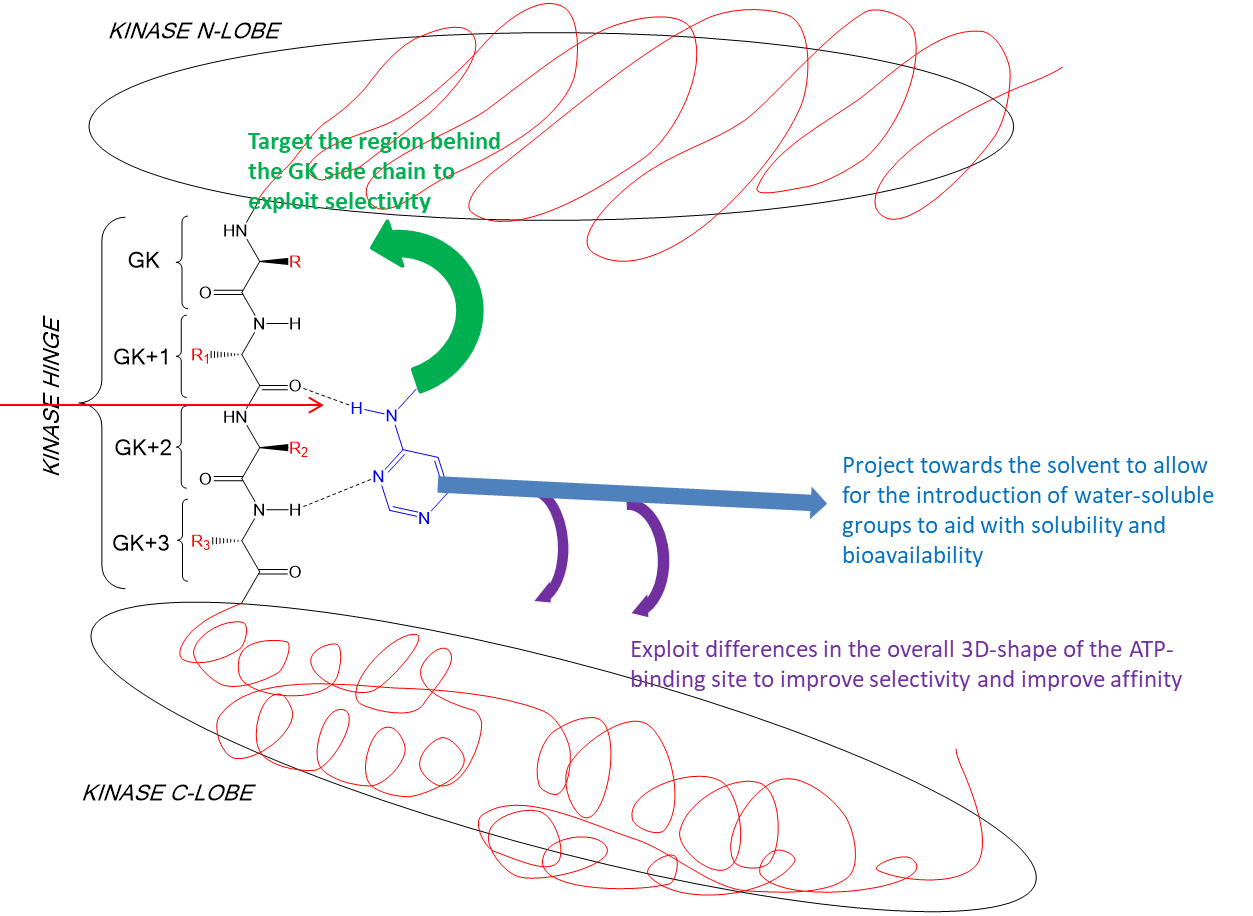
19: the first amino acid in the hinge region at the N lobe of the kinase is known as the gatekeeper (GK residue) as the R side chain is gatekeeper to a potential kinase selectivity pocket - this pocket can be exploited in drug development
The ATP adenosine head is bound to AAs by specific H bonds with the backbone of the GK+1 and GK+3 amino acids of the hinge region.
2 Mg ions bound by the kinase coordinate with ATP terminal phosphate to make it more reactive towards transfer to substrate.
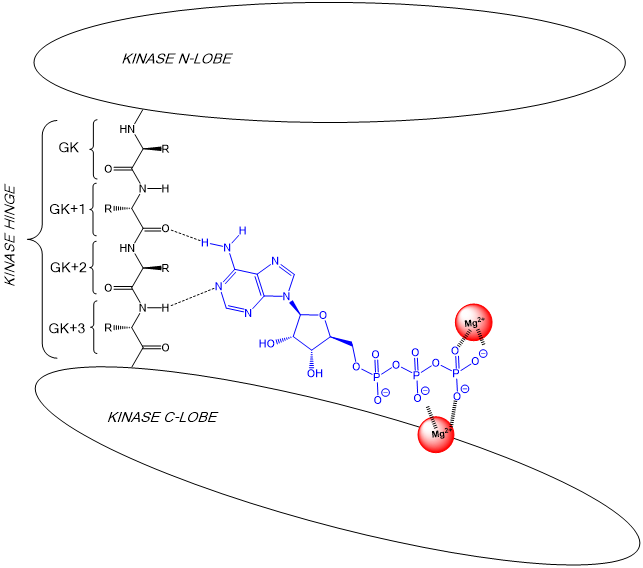
20: Substrate positions hydroxyl group of serine, threonine or tyrosine amino acid next to ATP’s activated terminal phosphate allowing phosphate transfer by nucleophilic substitution.
Substrate seen below in light blue
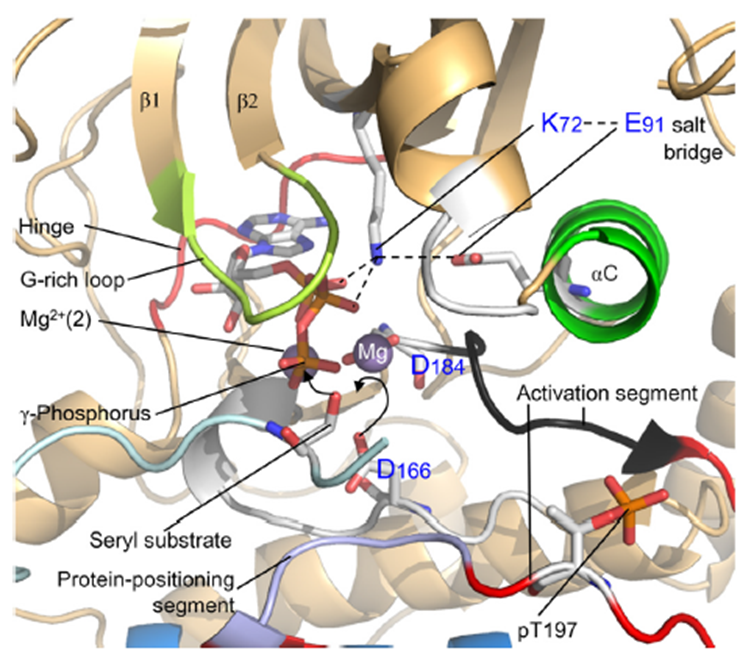
21:
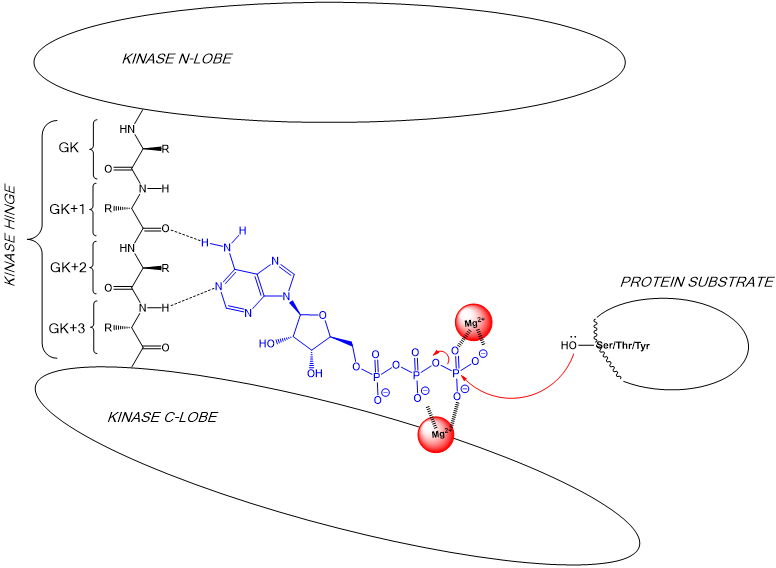
some kinases have dual specificity
22: The phosphorylated protein substrate then disengages from the kinase to leave and transmit the signal by interacting with another protein or proteins.
23: Kinase is now bound to cofactor ADP rather than ATP. ATP is released by the hinge opening and a new ATP binds so phosphorylation can continue when a new substrate molecule engages.
25: We can stop a kinase from transferring phosphate to its substrate by designing a molecule which competes with ATP for the binding site.
26: A molecule competing with ATP to bind a kinase must bind with higher affinity than ATP to compete it and therefore needs to make MORE interactions with the binding site to improve affinity eg ionic, hydrogen bonds, van der Waals, pi-cation, aromatic ring stacking interactions or aromatic ring edge to face interactions.
27: All 518 kinases have an ATP binding site so it must be made sure that the designed molecule is selective for only the specific kinase binding site, or a very minimal select number from the kinome
28: need to understand what amino acids present in the hinge region from gatekeeper onwards in order to make sure molecule only targets required kinase
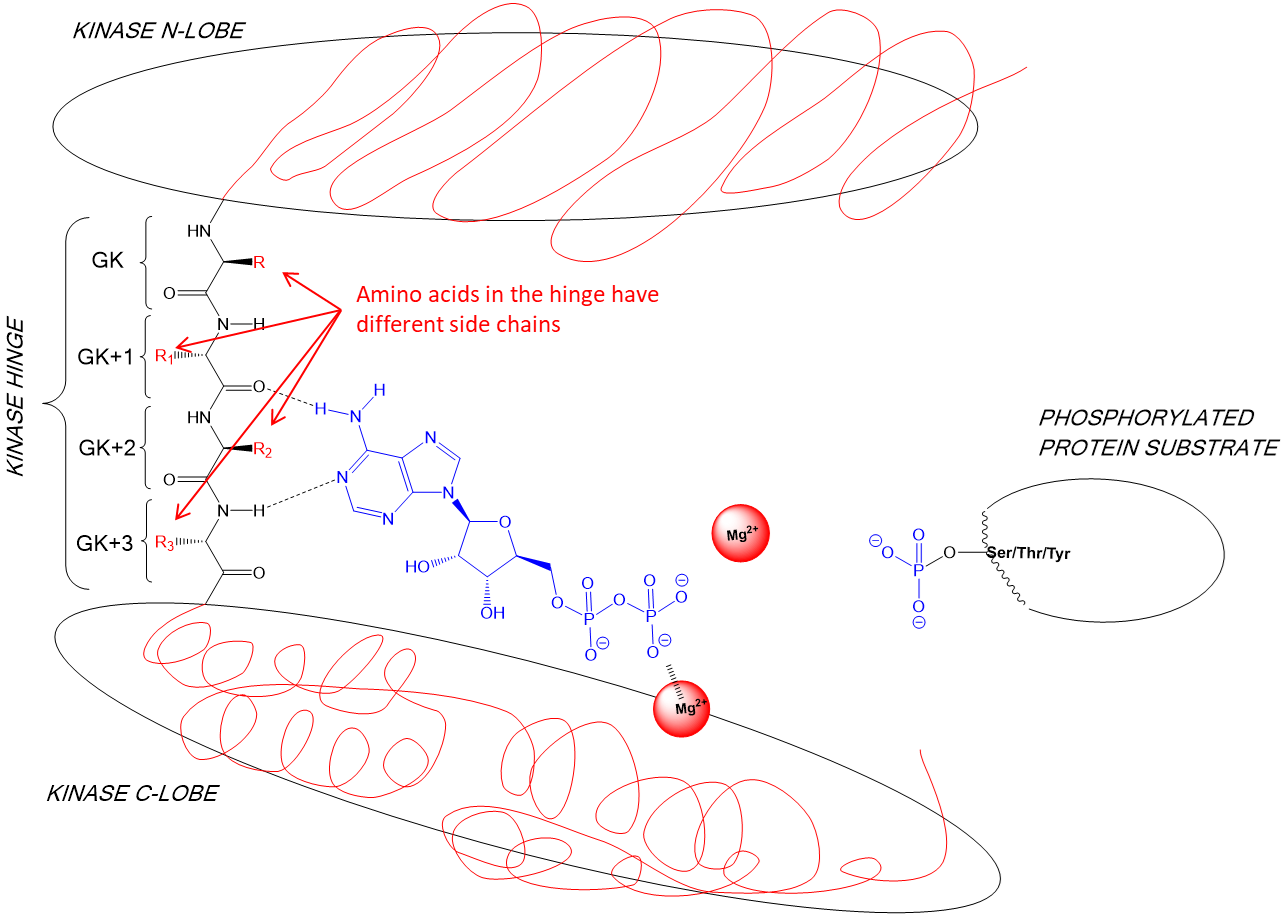
29: Amino acids in the N and C lobe have different side chains
30: shown is the variation within the CDK (cyclin dependent, involved in cell cycle) family of kinases

31: If ATP binding sites have different shapes then different molecules can be designed to map onto those different shapes. The surfaces below represent different sized amino acids present in kinase ATP binding sites
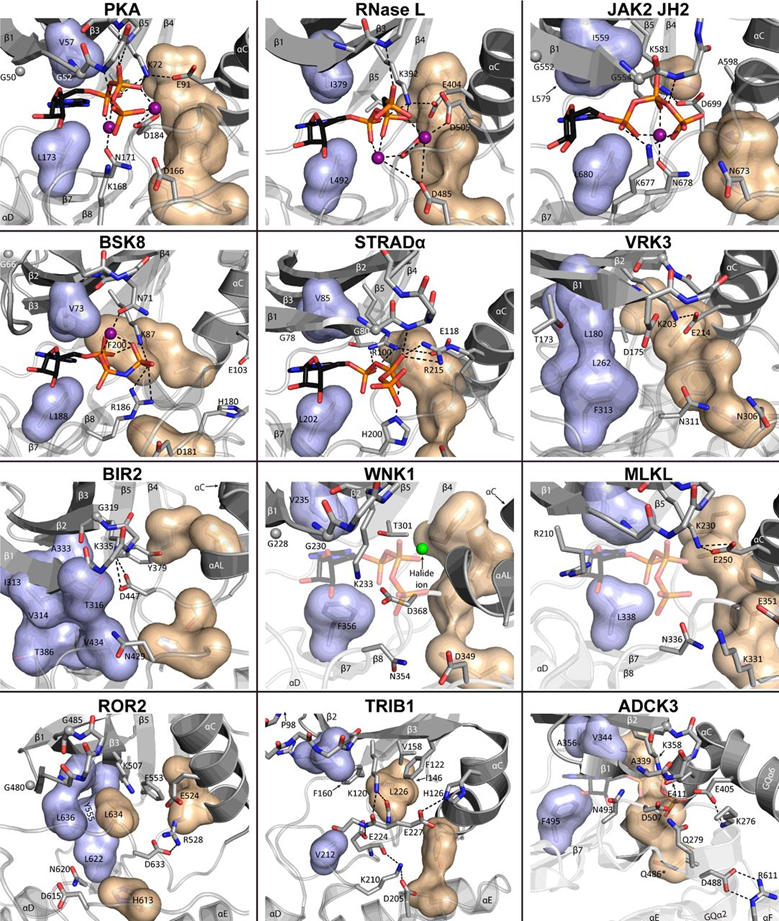
32: It’s not always possible (sometimes not even desirable) to achieve complete selectivity for a single kinase
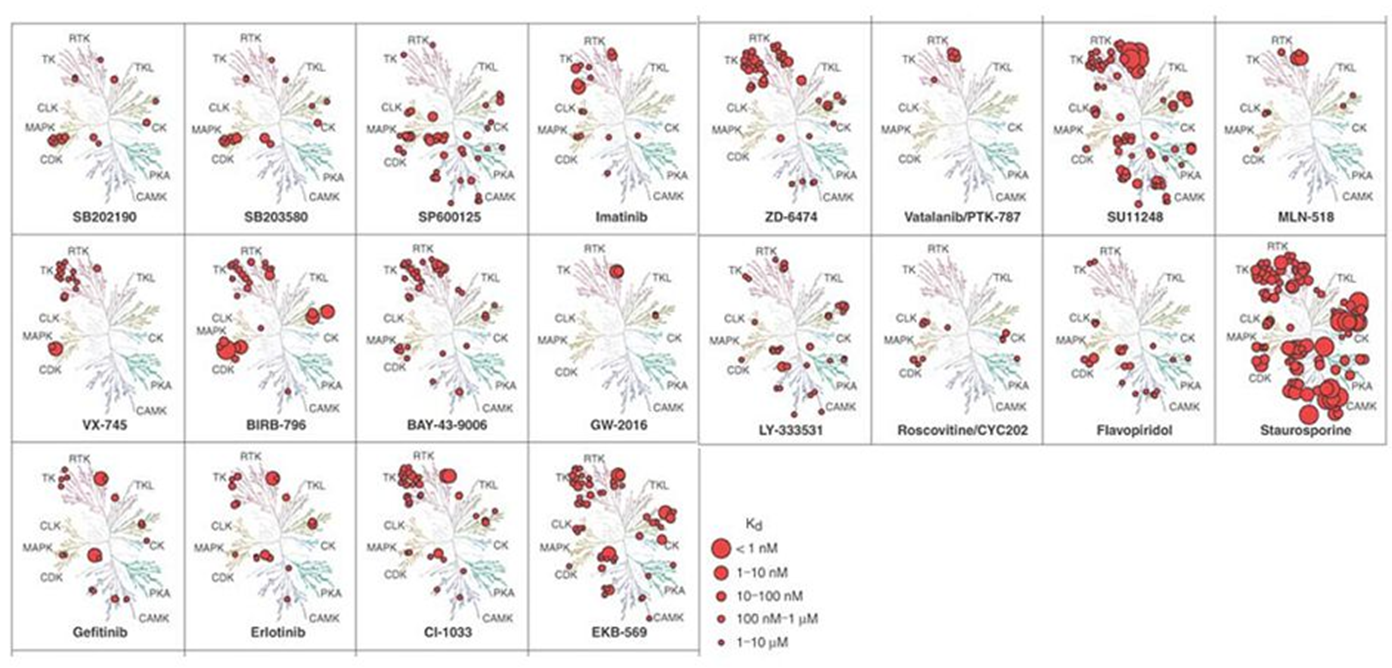
Below is way of representing kinase tree that shows how different inhibitors affect them Some eg staurosporine inhibit many kinases - used as positive control in kinase assays
Sometimes having an inhibitor with dual activity is beneficial eg could target multiple pathways in cancer
35: Kinase inhibitors usually begin as a heterocycle which targets the hydrogen bonding groups of the hinge region (shown in blue)
36: need to see if there’s a specific order/if different components of heterocycle have specific roles
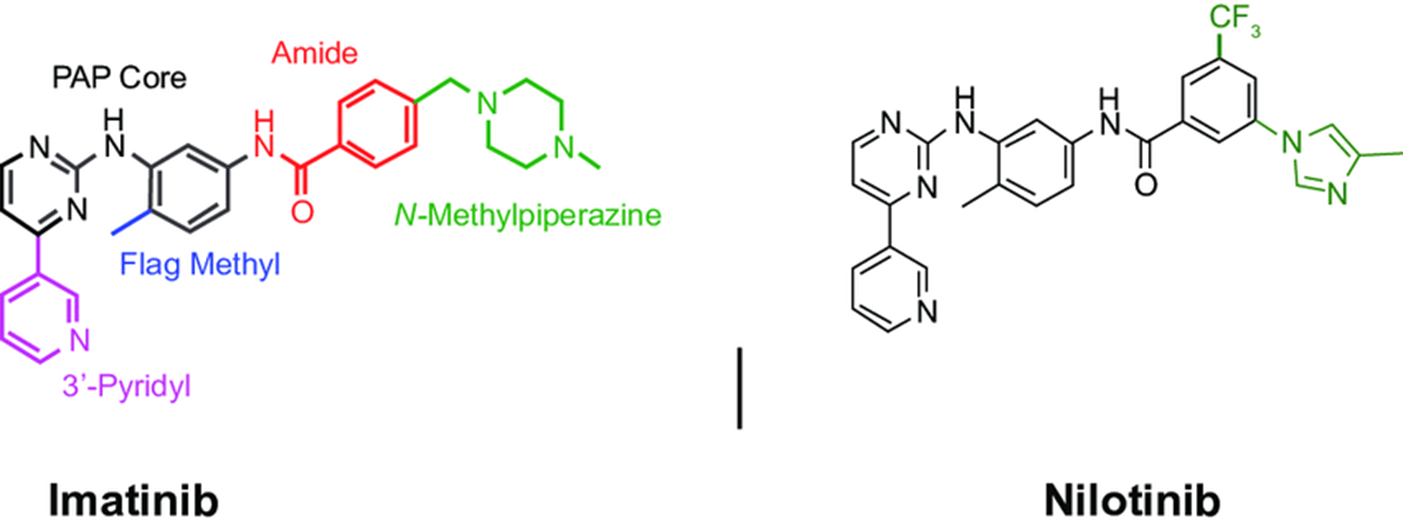
other structures then built onto the heterocycle to improve properties - modifications added in shown below
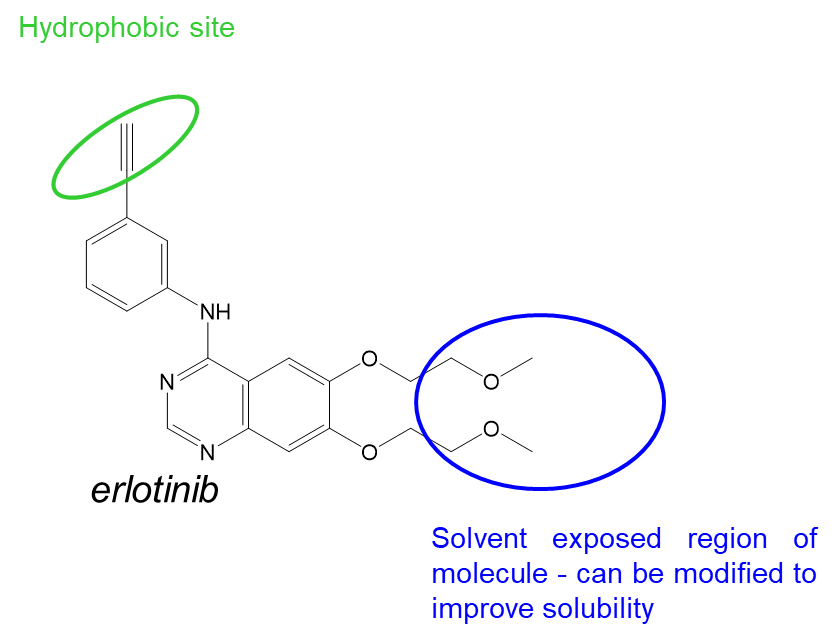
37: Gefitinib is a kinase inhibitor which targets EGFR. It competes with ATP by forming competitive interactions - hydrogen bonds to GK+3 (met 793) and targets the region behind the GK side chain (N lobe), Thr 790. It is a potent inhibitor with poor solubility and low bioavailability and is rapidly metabolised.
38: Design of gefitinib to target EGFR

39: Design of erlotinib to target EGFR - similar core structure/scaffold to gefitinib but designed independently by different company
40: Inhibitors imatinib and nilotinib were designed to target Bcr-Abl (formed by chromosomal translocation) in chronic myeloid leukaemia (CML). When not being inhibited, bcr-abl drives CEBP alpha mediated transcription which leads to cell growth.
Bcr-Abl treatment must be continued throughout patient life, resistance develops over time as imatinib loses its affinity for Bcr-Abl due to protein mutation
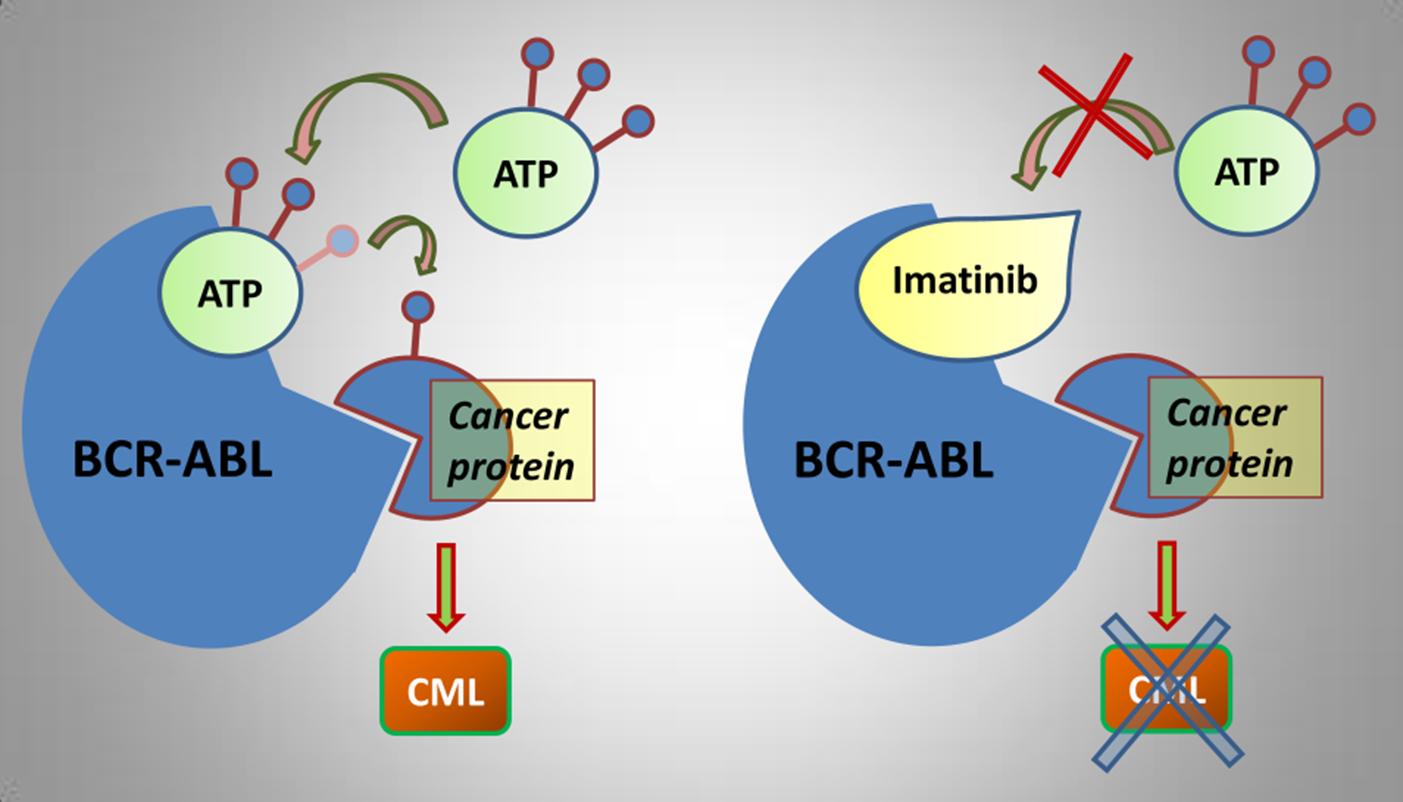
41: Imatinib and nilotinib hydrogen bond to GK +3 of bcr-abl hinge (Met 318) and hydrogen bond to the GK side chain (Thr 315). Moderately potent (5 micromolar) but has poor selectivity for Tyr kinases over Ser/Thr kinases (however was still able to compete for ATP), poor solubility and low bioavailability.
42: modified to add in flag methyl group which forces bond rotation. The amide group is now able to hydrogen bond with Glu 286 in the N lobe and Asp 381 in the C lobe, increasing affinity by 0.1 micromolar (50 fold increase) and increasing selectivity for Bcr-Abl and cKit (?). However the inhibitor still has poor solubility and low bioavailability
43: The inhibitor is further modified by addition of a tertiary amine making the inhibitor able to be formulated as a water soluble hydrochloride salt.
44: nilotinib = later generation
Extra reading:
See Patrick Chapter 21: Anticancer Agents – Inhibitors of the Abelson tyrosine kinase