U7 WBC
DISORDERS OF WHITE BLOOD CELLS AND LYMPHOID TISSUES
Covering leukocytes, lymphoid tissue, hematopoiesis, hematopoietic growth factors, nonneoplastic and neoplastic disorders of white blood cells, and plasma cell dyscrasias.
Hematopoietic and Lymphoid Tissues
Function: Protect the body against invasion by foreign agents.
Location of Blood Cell Formation:
Blood cells are formed in the myeloid (bone marrow); circulate, mature, and function in lymphoid tissues including: lymph nodes, thymus, and spleen.
Development of Blood Cells:
Involves interactions among precursor bone marrow cells, growth factors, cytokines (chemical messengers), and gene products like transcription factors
Leukocytes (White Blood Cells)
Definition: White blood cells (WBCs) include granulocytes (neutrophils, eosinophils, basophils), monocytes/macrophages, and lymphocytes.
Origin of White Blood Cells:
Granulocytes and agranular monocytes originate from the myeloid stem cell in the bone marrow, circulate in the blood.
T lymphocytes (T-cells) and B lymphocytes (B-cells) originate from lymphoid stem cells in bone marrow, and migrate between the blood and lymphatic system.
T Lymphocytes: Mature in the thymus; differentiate into helper T-cells and cytotoxic T-cells.
B Lymphocytes: Mature in bone marrow (myeloid) Differentiate into immunoglobulin-producing plasma cells.
Natural Killer (NK) Cells: Large granular lymphocytes that do not share specificity with T or B lymphocytes but can lyse target cells
Bone Marrow and Hematopoiesis
Hematopoietic System:
Arises from a small number of pluripotent stem cells (can-do-anything).
Supports development of hematopoietic precursors:
Erythroid → RBC
Myelocyte → granulocyte and monocyte
Lymphocyte → T-cell and B-cell
Megakaryocyte → platelets.
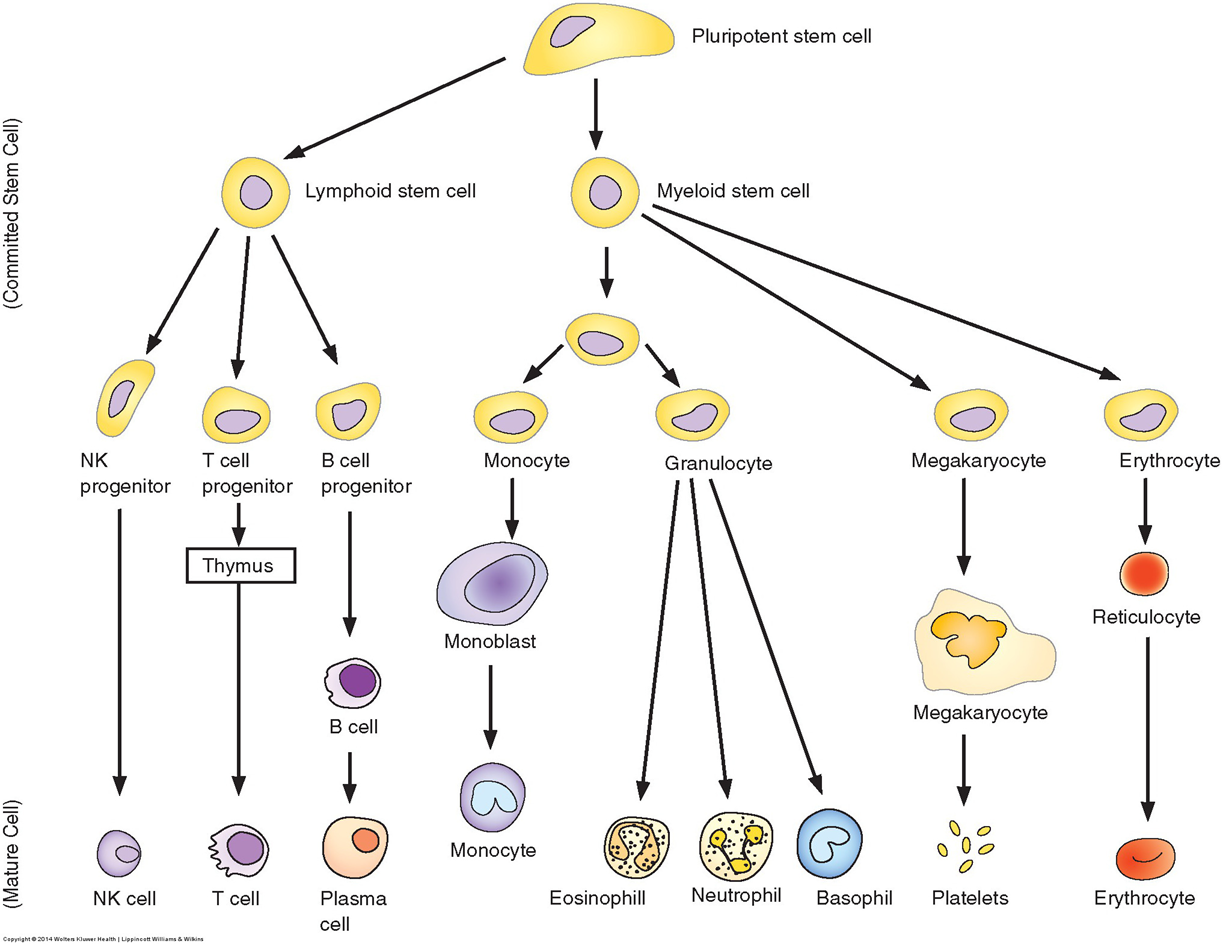
Unipotent Cells:
Progenitors of specific blood cell types.
B and T Lymphocyte Development:
Begins in bone marrow, ends in peripheral lymphoid structures (lymph nodes, thymus, and spleen); initial immune response via neutrophils lasts 4-5 days
Hematopoietic Growth Factors
Function: Control leukopoiesis (production of WBCs).
3 Types of Hematopoietic Growth Factors
1.Specific cell lineage development growth factors.
2.Factors affecting early multipotential progenitor cells.
3. Factors inducing growth factor gene expression in other cells
Leucocyte Developmental Stages
Lifespan of WBCs: Short; continuous renewal crucial for normal blood levels.
Leucocyte Development: Begins with myeloid / lymphoid stem cells in bone marrow and categorized as blast cells→ immature precursors
§Myeloid stem cells → granulocyte and monocyte cell lines
lymphoid stem cells → lymphocytes
Key Characteristics of WBC Disorders: Conditions can decrease stem cell or hematopoietic growth factor availability leading to lower WBC counts
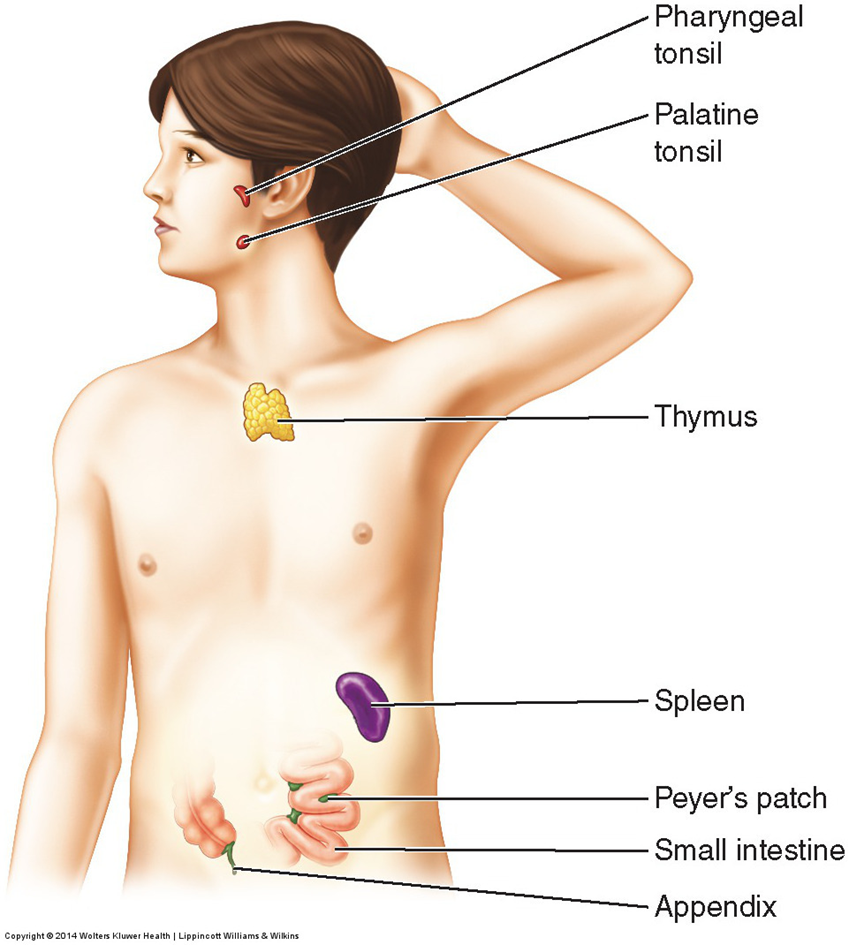
Lymphoid Tissues
Components: Lymphatic system composed of lymphatic vessels, lymphoid tissue, lymph nodes, thymus, and spleen.
Development of B and T Lymphocytes: Both start in bone marrow, with:
B lymphocytes differentiating into plasma cells, proliferating and producing antibodies in lymph nodes.
T lymphocytes become helper T-cells and cytotoxic T-cells in the lymph tissues
Lymph Nodes:
Organized lymphoid tissue along lymphatic vessels, varying from 1 mm to 1-2 cm in size, with a fibrous structure supporting a delicate reticular network.
Parenchyma divided into outer cortex with defined B and T cell domains and a medulla.
Lymphomas can arise from lymph nodes and MALT → not incased lymph tissues
Nonneoplastic Disorders of White Blood Cells
Physiologic Number of Leukocytes: 5-10,000 cells/µL in peripheral circulation.
Types of Disorders:
Leukopenia (deficiency of leukocytes)
Proliferative disorders with increased leukocyte numbers (leukocytosis).
Chemotherapy-induced leukopenia affects neutrophils primarily
Neutropenia (Agranulocytosis) <1000/ul
Definition: Decrease in absolute neutrophils, defined as circulating count < 1000/µL.
Causes:
Can arise from decreased production, accelerated utilization, or shifts from blood to tissue compartments.
Key Points: Degree of neutropenia correlates with increased infection risk, especially during severe infections (all used up, can’t make more neuts fast enough).
In the absence of infection, neutropenia is associated with immunologic disorders
Types:
Congenital→ alloimmune neonatal neutropenia
Acquired: autoimmune conditions, hematologic malignancies, etc.
Severe Cases:
Aplastic anemia leads to anemia, thrombocytopenia, and agranulocytosis
vSevere neutropenias:
§Pancytopenia: condition when all the RBCs, WBCs, and platelets are considerably low
§Agranulocytosis: characterized by the virtual absence of neutrophils
§In aplastic anemia, the affected individual has a depletion of all the myeloid stem cells → anemia, thrombocytopenia, and agranulocytosis
Infectious Mononucleosis
Definition: Self-limiting, lymphoproliferative syndrome caused by acute viral infection of B-lymphocytes.
Causative Agent: Epstein-Barr virus (EBV) accounts for 85-90% of IM cases; CMV accounts for the rest.
Transmission: Primarily through saliva (“kissing disease”) and other body fluid secretions.
Symptomatic IM: Common among adolescents and young adults (ages 15-35), with peak incidence at 15-24 years; rare after 40 but more often caused by CMV.
Clinical Manifestations: Pharyngitis, lymphadenopathy, fever; onset of sore throat and fever; classic symptoms of EBV-induced IM include fever lasting 7-10 days, sore throat, cervical lymph node tenderness.
Complications:
About 50% of cases exhibit splenomegaly x2-3 normal size. Immune mediated hepatitis.
Treatment: Usually self-limiting; management is symptomatic
Neoplastic Disorders of Lymphoid and Hematopoietic Origin
Overview: Neoplasias of lymphoid origin may arise from B or T cells and encapsulate various stages of development. Major categories being non-Hodgkin lymphomas (NHLs), Hodgkin lymphoma, lymphoid leukemias, and plasma cell dyscrasias.
Clinical Features: Determined by cell origin and molecular events driving transformation to malignancy. Neoplasm disseminates from onset d/t blood circulation.
Malignant Lymphomas
Definition: Diverse group of solid tumors comprised of neoplastic lymphoid cells.
Characteristics:
Tumors can arise from primary (thymus/bone marrow) or secondary lymphoid tissue (lymph nodes, spleen, intestinal lymphoid tissues etc.) and vary across genetics and treatment response.
Causes: Often initiated by genetic mutations and viral infections, commonly affecting lymph nodes or gastrointestinal lymphoid tissues
vTwo (2) major types of malignant lymphomas are Hodgkin lymphoma (HL) and non-Hodgkin lymphoma (NHL)
§Children are less likely to be diagnosed with NHL
vLymphoma incidence rates vary with respect to age, assigned-at-birth sex, geographic location, and socioeconomic class
Non-Hodgkin Lymphomas (NHL)
NHL Definition: Clinically diverse group arising from B-cells, T-cells, and NK cells,
Responsible for approximately 25% of new cancer cases in the US. Highly common in older adults.
Incidence: Has significantly increased since the 1970s; variations remain related to environmental and genetic factors.
NHL Classification:
Divided into B-cell neoplasms (including myelomas)
T-cell/NK-cell neoplasms.
vNon-Hodgkin lymphomas arise from the lymphocytes
§Site of origin varies according to their common lymphoid progenitor
§Exact etiology of the alteration is unknown
§Linked pathogens: EBV, human T-lymphotropic virus 1, and H. pylori have been linked to specific subtypes
Affected Sites: Cervical, axillary, inguinal, and femoral lymphatic chains where painless lymph node swelling and transformation occurs, with systemic manifestations such as night sweats and weight loss. Effects immunosuppressed, older people. Must be confirmed by a lymph node bx.
Mature B-Cell Lymphomas
Follicular lymphomas:
§Peak incidence, age 60
§↑ risk of secondary malignancies for > 65 y/o at the time of dx
§Lymph nodes are usually affected
oOther areas of frequent involvement: spleen, bone marrow, peripheral blood, head and neck regions, GI tract, and skin
§Over time, ≈ 33% of follicular lymphomas transform into fast-growing diffuse large B-cell lymphomas
vDiffuse large B-cell lymphomas:
§Heterogeneous group of aggressive germinal or post-germinal center neoplasms
§Occur in all age groups; most prevalent between 60 and 70 years
§Etiology unknown, but may involve EBV or HIV infection
§Rapidly evolving, multifocal, nodal, and extranodular tumors
§Quickly fatal if untreated
Burkitt lymphoma:
§Disorder of germinal B cells
§Fastest-growing human tumor: accounts for 30% of childhood lymphomas worldwide
§Most aggressive and rapidly growing of the NHLs
§Endemic to areas of Africa where both Epstein-Barr virus and malaria are common
Characterized by a rapidly growing tumor primarily affecting adversely the jaw and facial features
Hodgkin Lymphoma (HL)
Hodgkin Lymphoma Definition: Specialized lymphoma distinguished by Reed-Sternberg cells
Bimodal age distribution peaks in early adults and older adults.
§Early adults (15-40 y/o)
§Older adults ( ≥ 55 y/o)
Key Differences From NHL: Presents in a singular node (or chain of nodes) compared to NHL's tendency for extranodal primary sites. Presence of Reed-Sternberg cells is diagnostic→ Characterized by large, atypical, multinucleated tumor cells
Etiology: Unknown, but genetic and environmental factors (radiation exposure, viral infections) may contribute
Clinical Manifestations: Enlarged, painless lymph nodes above the diaphragm; mediastinal masses common→ cough, dyspnea
Systemic symptoms such as intermittent fevers and pruritus can occur, night sweats
fatigue→ disease spread
Definitive diagnosis via biopsy
Nodular lymphocyte-predominant HL
§Represents only a small portion of the cases
§Unique feature: generally exhibits a nodular growth pattern
oWith or without diffuse areas and rare Reed-Sternberg cells (“popcorn” -- lymphohistiocytic cells)
§Often localized rather than disseminated at the time of dx
§Slowly progressive course
§Survival rate > 80%
Classic HL
§Clonal proliferation of typical mononuclear Hodgkin cells and multinucleated Reed-Sternberg cells with invariable expression of CD30 (cell surface marker)
§Four (4) variants of classic HL:
i. Nodular sclerosis ~ most common and often found in adolescents and young adult assigned-at-birth females, 15-35 y/o
ii. Mixed cellularity
iii. Lymphocyte-rich (newly defined entity)
iv. Lymphocyte-depleted (rarely dx)
Leukemias
Leukemia Definition: Malignant neoplasms of cells arising from hematopoietic precursor cells, characterized by unregulated proliferation, leading to overcrowding of bone marrow. Can spill into other organs like liver, spleen, lymph nodes.
Types: Classified based on predominant cell of origin (myeloid or lymphoid) and rate of progression (acute or chronic). Four main types include:
Acute lymphocytic leukemia (ALL)
Acute myelogenous leukemia (AML)
Chronic lymphocytic leukemia (CLL)
Chronic myelogenous leukemia (CML)
Epidemiology: Higher incidence in males; prevalent in children compared to adults and known to account for a significant number of cancer diagnoses.
vLeukemias are the most frequent cancer etiology in children and adolescents
§Leukemia accounts for 28% of childhood cancers
§Acute lymphoblastic leukemia (ALL) is the least overall common leukemia type; but the most common leukemia in children (≈ 66% of cases dx before age 20)
oLeukemias account for 34% of childhood cancers, and ALL accounts for 78% of new leukemia cases in children
vLeukemias are dx 10X more frequently in adults than in children
Pathogenesis: Unknown etiology; often linked to clonal disorders and genetic predispositions; chromosomal aberrations like the Philadelphia chromosome (translocations between 9 and 22) notable in CML
ACUTE LEUKEMIA
vAcute leukemias:
§Usually have a sudden onset; WBCs are poorly differentiated
§Seen in both at-birth-assigned sexes and in all ages; incidence ↑ dramatically after age 50; affected have a significantly lower survival rate than in chronic leukemias
§Frequent presenting symptomatology: fatigue, fever (low-grade), blurred vision, dyspnea, bone and joint pain, weight loss, pale integument, infections (persistent or frequent), bleeding diathesis (excessive bruising and bleeding)
§Key objective clinical manifestations: low-grade fever and high blast count
§The two (2) most common types of acute leukemias: acute lymphocytic leukemia (ALL); and acute myelogenous leukemia (AML)
Acute lymphocytic leukemia (ALL)
•Rare and aggressive
ü May affect the bone marrow, lymph nodes, spleen, integument, and central nervous system
•Occurs most frequently in children: 75% of all childhood leukemia cases
ü≈ 90% of the affected have numeric and structural changes in the chromosomes of their leukemic cells
•Unexpected manifestations: confusion, ascites, and pleural effusions,
•Frequent symptomatology: persistent flu-like symptoms, lymphadenopathy of unknown etiology, petechiae, minor hemorrhaging (frequent epistaxis, gingivitis)
•≈ 33% of ALL cases occur in adults; account for 80% of adult leukemia deaths
ü30-40% of affected adults achieve remission
Acute myelogenous leukemia (AML)
•Mainly a dz of older adults, but also seen in children and younger adults
üAccounts for 25% of cases in children and adolescents
•Diverse group of neoplasms affecting myeloid precursor cells in the bone marrow
•Associated pathologies: organomegaly (spleen and liver), central nervous system involvement (headaches, confusion, seizures or cranial nerve palsies)_
•AML is most associated with acquired genetic alterations inhibiting terminal myeloid differentiation
üSpecific chromosomal abnormalities, including translocations, are seen in a large number of AMLs
Clinical Manifestations: ALL and AML typically present with similar clinical features, including abrupt onset of symptoms
oFatigue 2°anemia
oLow-grade fever; high blast cell counts
oNight sweats
oWeight loss 2° rapid proliferation and hypermetabolism of the leukemic cells
oBleeding 2°↓ platelet count
oBone pain and tenderness 2° bone marrow expansion
oInfection 2° neutropenia ~ risk ↑ sharply as neutrophil count falls to < 500 cells/microliter
oGeneralized lymphadenopathy, splenomegaly, and hepatomegaly 2° infiltration of leukemic cells ~ more common in ALL
vLeukostasis: circulating blast count becomes markedly elevated, thus ↑ blood viscosity
§Predisposes to lymphoblastic emboli with associated obstruction of small blood vessels in the pulmonary and cerebral circulation
vHyperuricemia: occurs due to the proliferation or ↑ breakdown of purine nucleotides (one of the components of nucleic acids) 2°leukemic cell death resulting from chemotherapy
§Can also occur before or during tx
vDiagnosis:
§Based on blood and bone marrow studies: leukemic cells in the peripheral blood, bone marrow, or extramedullary tissue
§Blood assays reveal the presence of immature WBCs in the circulation and the bone marrow constituting 60-100% of the cells
oAnemia is almost always present
oPlatelet count is ↓
oBlast cells (immature blood cells that develop from stem cells in the bone marrow) are often abnormal and multiply rapidly → ↑ blast cell counts
§Cytogenic studies are used to detect chromosomal abnormalities: among the most powerful prognostic indicators
CHRONIC LEUKEMIA
vChronic leukemias:
§Proliferation of more fully differentiated myeloid and lymphoid cells (than the immature cells found in the acute leukemias)
§Account of the majority of adult leukemias
§Two (2) most common types: chronic lymphocytic leukemia (CLL); and chronic myelogenous leukemia (CML)
oSeveral forms of CML can occur, depending on the lineage of the malignant cells
§Mostly a disorder of older adults
oIncidence ↑ after age 40: mean age at CML and CLL dx ≈ 70
oRare in those < 40 y/o; infrequent in children
§Chronic leukemic cells are well-differentiated; the dz process advances slowly and insidiously; and affected individuals have a longer life expectancy
§Key clinical manifestations: hx weakness, fatigue, and weight loss
Chronic lymphocytic leukemia (CLL):
•Clonal malignancy of B lymphocytes
•Most common adult leukemia in the Western world
•Number of genomic alterations ↑ throughout the course of the dz
•Immunotyping confirms the dx
•Some affected individuals survive for many years without tx; others have fatal dz despite aggressive tx
oMore aggressive CLL forms experience a more rapid sequence of clinical deterioration characterized by
•↑ lymphadenopathy
•Hepatosplenomegaly
•Fever
•Abdominal pain
•Weight loss
•Progressive anemia
•Thrombocytopenia with a rapid ↑ in the lymphocyte count
vDiagnosis:
§Chronic lymphocytic leukemia (CLL)
oDiagnostic hallmark is isolated lymphocytosis
oWBC is usually > 20,000/µL; may be elevated to several hundred thousand, usually 75-98% are lymphocytes
oHct and lymphocyte counts are usually within defined limits at the time of dx
oAffected individuals may retain normal plasma synthesis and fully differentiated myeloid and lymphoid cells= chronic lymphocytic leukemia
Chronic myelogenous leukemia (CML):
•Disorder of pluripotent hematopoietic progenitor cells
•Characterized by excessive proliferation of marrow granulocytes, erythroid precursors, and megakaryocytes
•CML cells have a distinctive cytogenic abnormality, the Philadelphia chromosome
Clinical Manifestations: Onset is usually slow, with presenting nonspecific symptoms such as weakness, fatigue, and weight loss (for a period of ≥ 6 months)= chronic myelogenous leukemia
üSplenomegaly often → a sensation of abdominal fullness and discomfort
üHepatomegaly is less common
•Lymphadenopathy is relatively uncommon
oAffected individuals can remain in the non-accelerated phase (chronic CML) for years before the dz process accelerates and culminates in a crisis
Diagnosis
oDiagnostic hallmark is an elevated WBC with a median count of 150,000/µL
•In some cases, WBCs are only moderately elevated
oDz hallmark: the presence of BRC-ABL (breakpoint cluster region-Abelson leukemia virus) gene product (detected in the peripheral blood)
oBone marrow examination is usually not necessary
•Helpful for prognosis and detecting additional chromosomal abnormalities
Plasma Cell Dyscrasias
Definition: Characterized by an increase in serum levels of a single monoclonal immunoglobulin or its fragments resulting from expansion of a single clone of immunoglobulin-producing plasma cells.
Types: Include multiple myeloma (MM), localized plasmacytoma, lymphoplasmacytic lymphoma, amyloidosis, and monoclonal gammopathy of undetermined significance (MGUS).
MGUS is seen as a premalignant condition closely linked to multiple myeloma
Multiple Myeloma (MM)
Overview: B-cell malignancy, most frequent and aggressive plasma cell tumor.
Etiology: Unknown, but may involve genetic factors and chronic stimuli from environmental sources; prevalence increases with age, with a median age of 71 years for diagnoses.
vPathogenesis:
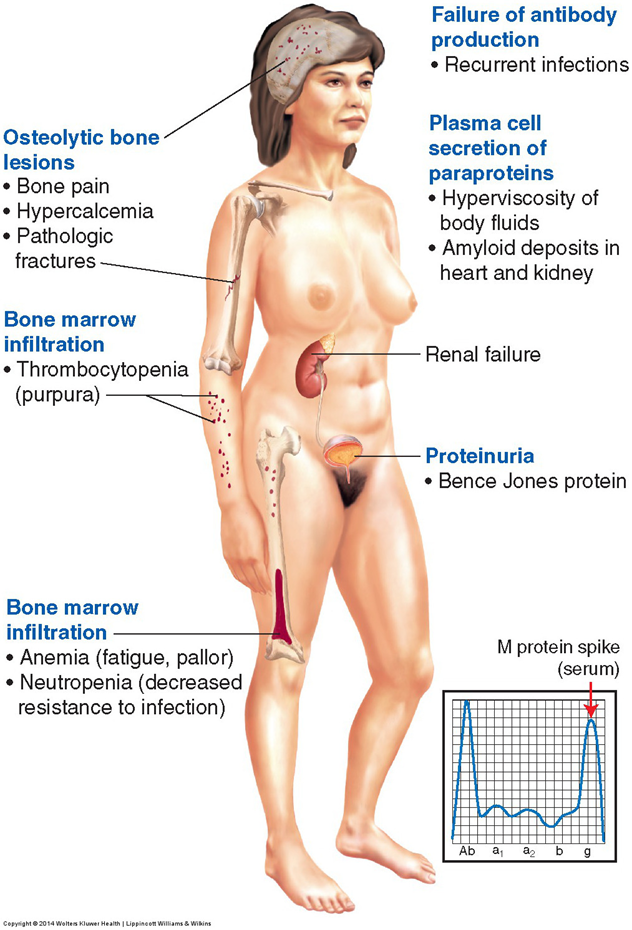
§Clonal plasma cell cancer characterized by slow proliferation of malignant cells as tumor cell masses scattered throughout the skeletal system and occasionally in soft tissue → bone destruction
Abnormal proliferation of plasma marrow cells; and proliferation and activation of osteoclasts
Precise etiology: unknown
oGenetic factors and chronic stimulation of the mononuclear phagocyte system by bacteria, viral agents, and chemicals → agent orange may play a role
oSlightly more common in assigned-at-birth males than in assigned-at-birth females
§Risk factors:
oModifiable: obesity, exposure to ionizing radiation, occupational exposure to herbicides and pesticides
oNon-modifiable: chronic immune stimulation, and autoimmune disorders
vMajor clinical manifestations:
§Main sites of involvement: bone and bone marrow
oReported symptoms: bone pain, fatigue
oSigns:
•Hx of bone fractures; bone lesions
•Elevated serum calcium (hypercalcemia)
•Elevated serum creatinine
•Weight loss, anemia, proteinuria
•Recurrent infections 2° suppression of the humoral response
•Renal dz/failure (occurs in ≈ 50% of those affected ~ see Fig. 24.14)
vChemotherapy is the tx of choice