Urine Screening for Metabolic Disorders
URINE SCREENING FOR METABOLIC DISORDERS
Overflow Versus Renal Disorders
Overflow Disorders
Involves disruption of normal metabolic pathways.
Increased plasma concentrations of nonmetabolized substances.
Renal tubules' ability to reabsorb is overridden.
Often results from inherited lack of specific enzymes, also known as inborn errors of metabolism.
Renal Disorders
Result from malfunctions in tubular reabsorption mechanisms.
Disorders Classified by Defect
Overflow Inherited Disorders
Overflow Inherited Disorders
Phenylketonuria
Tyrosinemia
Alkaptonuria
Maple syrup urine disease
Organic acidemias
Cystinosis
Porphyria
Mucopolysaccharidoses
Galactosemia
Lesch-Nyhan disease
Metabolic
Infantile Tyrosinemia
Melanuria
Indicanuria
5-Hydroxyindoleacetic acid
Porphyria
Renal
Hartnup disease
Cystinuria
Phenylalanine/Tyrosine Metabolic Pathway
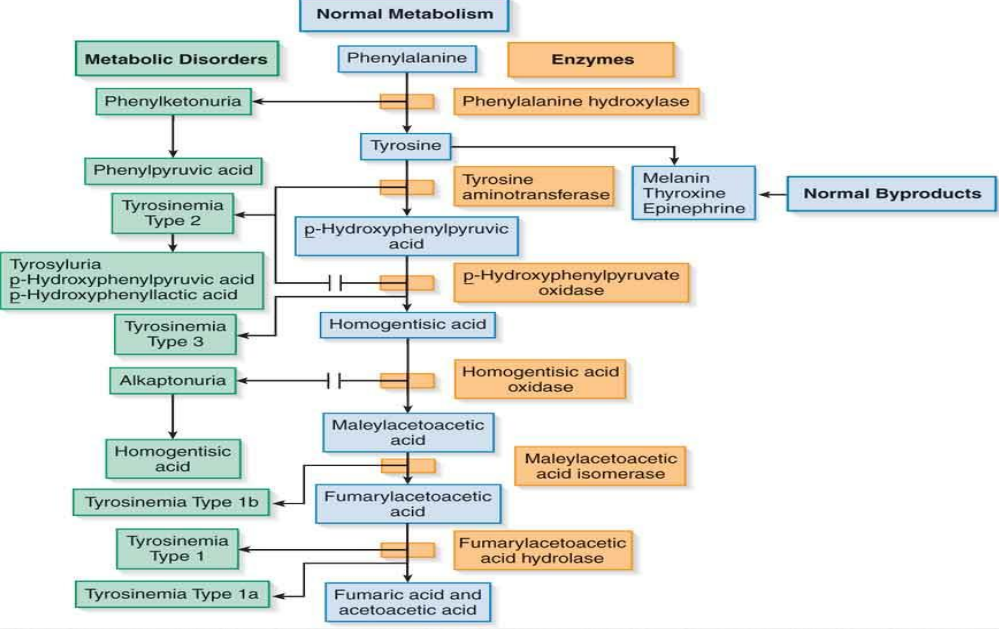
Amino Acid Disorders (Aminoacidurias)
Phenylketonuria (PKU)First identified in 1934 with a characteristic mousy odor in urine.Caused by an inability to produce the enzyme phenylalanine hydroxylase.Management involves strict dietary exclusions of phenylalanine, particularly from milk products.Consequences include developmental issues due to accumulated phenylalanine.
Ferric Chloride Tube TestPurposeA urine test specifically targeting phenylpyruvic acid.ProcedureTransfer 1 mL urine into a test tube.Add 5 drops of 10% ferric chloride.InterpretationPositive Result: Permanent blue-green color indicates phenylpyruvic acid presence.
Guthrie TestOverviewThe most established test for PKU detection.ProcedureBlood from a heel stick is applied to filter paper disks.Disks are incubated in culture media containing beta-2-thienylalanine.InterpretationPositive Result: Growth of organism confirms PKU presence.
Tyrosyluria/TyrosinemiaDeficiency CausesEnzyme deficiencies leading to tyrosine disorders:
Type 1: Fumarylacetoacetate acid hydrolase deficiency leading to severe renal and liver dysfunction.
Type 2: Deficiency in tyrosine aminotransferase causing specific symptoms like corneal erosion.
Type 3: Deficiency of p-hydroxyphenylpyruvate oxidase associated with mental retardation.
Types of Tyrosinemia
Type 1: Severe deficiency leading to acute manifestations in infants.
Type 2: Involves visual symptoms and skin lesions.
Type 3: Less common; correlates with cognitive challenges when not under dietary restrictions.
MelanuriaOverviewRelated to a secondary pathway for tyrosine involving:Production of melanin, thyroxine, epinephrine.Associated ConditionsIncreased production can signal malignant melanoma, leading to detectable compounds in urine.
AlkaptonuriaPathophysiologyArises from a deficiency in homogentisic acid oxidase.Characterized by dark urine (alkaline), leading to potential liver and heart diseases later.
Urine Color Reactions
Ferric chloride test: Produces transient deep blue coloration.
Clinitest: Yields yellow precipitate.
Silver nitrate and ammonium hydroxide: Leads to black coloration.
Branched Chain Amino Acid DisordersTypes
Maple Syrup Urine Disease (MSUD): Elevated levels of leucine, isoleucine, and valine.
Organic Acidemias: Includes isovaleric, propionic, and methylmalonic acidemias.
Maple Syrup Urine Disease (MSUD)OverviewCharacterized by honey maple odor in urine, appearing after one week.Dietary regulation needs to begin by day 11 for favorable outcomes.
2,4-Dinitrophenylhydrazine (DNPH) TestProcedureAdd 1 mL of urine to the test tube.Introduce 10 drops of 0.2% DNPH in 2N HCl.Wait for 10 minutes; observe for precipitate.
Organic AcidemiasGeneral SymptomsSymptoms include:
Vomiting
Metabolic acidosis
Hypoglycemia
Ketonuria
Increased serum ammoniaSpecific Types
Isovaleric, propionic, methylmalonic acidemias.
Isovaleric, Propionic, Methylmalonic AcidemiasSymptoms and Mechanisms“Sweaty feet odor” linked to isovaleric acidemia.Propionic and methylmalonic acidemias characterized by halted conversion of amino acids to the necessary coenzyme.
Tryptophan DisordersMetabolites InvolvedIncreased urinary excretion of indican and 5-Hydroxyindoleacetic acid (5-HIAA).Tryptophan conversion involves liver processes; abnormalities cause excess in urine and feces.
IndicanuriaMechanismTryptophan overlaps between intestinal reabsorption and conversion to indole by gut bacteria.Increased indole is processed in the liver, converting to indican for renal excretion.
Hartnup DiseaseSymptomsKnown as blue diaper syndrome that affects intestinal reabsorption leading to specific symptoms.
5-Hydroxyindoleacetic Acid (5-HIAA)ProductionSerotonin produced from tryptophan in the intestinal argentaffin cells has systemic implications regarding tumors.
Urine Test for 5-HIAATesting ProcedureNitrous acid reacts producing a purple-black coloration.Normal range is 2 to 8 mg/day; anything > 25 mg/day indicates disease.
Cystine DisordersTypes
Cystinuria: Inherited disorder affecting renal reabsorption.Increased sulfur odor characteristic of both disorders.
Cystinuria TestsTesting ProcedureCyanide-nitroprusside test produces a noticeable red-purple color if high cystine is present.False positives can arise from several conditions including ketonuria and homocystinuria.
Cyanide-Nitroprusside TestStepsAdd sodium cyanide to urine.Wait some duration before adding nitroprusside.Observe results for color change.
CystinosisOverviewGenetic disorder leading to deposits in tissues from defective lysosomal transport mechanisms.Deposits lead to serious complications including renal failure over time.
Cystinosis CategoriesTypes
Nephropathic: Rapid progression, severe outcomes.
Non-nephropathic: Benign with occasional ocular issues.
HomocystinuriaSymptoms and ManagementCaused by metabolism defects of methionine leading to serious health issues.Dietary modifications are essential for management.
Silver Nitroprusside TestTest ProcedureCombine urine with ammonium hydroxide.Introduce silver nitrate and sodium nitroprusside.Observe for specific color changes.
Porphyrin DisordersOverviewInvolves disruptions in heme synthesis pathways, affecting various body fluids.Detection using multiple tests on urine, blood, and feces.
Heme SynthesisPathwaysDetail the biochemical routes for heme production, and how they correlate with symptoms.
Porphyria ClassificationCategories
Inherited: Genetic causes; presents with specific symptoms.
Acquired: Conditions leading to secondary porphyrin levels, such as lead poisoning.
Color and Fluorescence TestingColor ChangesPort wine coloration indicates potential porphyrin presence in fluid.Use of fluorescence examined to confirm results under UV exposure.
Mucopolysaccharide DisordersOverviewInherited conditions leading to non-metabolized polysaccharide accumulation.Specific substances detectable in urine provide diagnostic insight.
Specific DisordersTypes
Hurler syndrome: Severe mental retardation and structural anomalies.
Hunter syndrome: Similar to Hurler but sex-linked.
Sanfilippo syndrome: Primarily affects cognitive function.
Urinary Screening TestsTechniquesAcid-albumin and cetyltrimethylammonium bromide turbidity tests identify significant anomalies.
Cetyltrimethylammonium Bromide (CTAB) TestProcedureCombine urine with CTAB.Evaluate turbidity for results.
Mucopolysaccharide Paper TestTesting StepsPrepare filters with azure A dye.Observe for color reactions post urinalysis to determine presence.
Purine DisordersLesch-Nyhan DiseaseInherited disorder involving severe uric acid consequences leading to cognitive and physical complications.
Carbohydrate DisordersOverviewConditions frequently detected via urine, notably galactosemia.Associated with enzyme deficiencies leading to significant health challenges.
Other MelituriasFormsConditions like lactosuria and fructosuria can occur due to various physiological states.
ConclusionQuote"There is nothing so difficult to comprehend for someone who reads a lot" - E.C. Javier,RMT.