Macular Dystrophies 1
Stargardt Disease (Fundus Flavimaculatus)
Genetics / Epidemiology
Autosomal recessive in of cases; sporadic or other inheritance patterns rare.
Prevalence → technically a rare disease, yet the single most common hereditary macular dystrophy.
Usually manifests in childhood (often <10 yrs).
Pathophysiology & Key Features
Primary degeneration of the retinal pigment epithelium (RPE) with secondary photoreceptor loss.
Lipofuscin (toxic bis-retinoid by-product of the visual cycle) accumulates in RPE and photoreceptors.
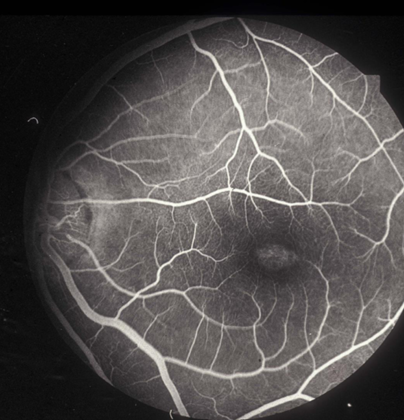
Characteristic “silent (dark) choroid” on fluorescein angiography (blocked background choroidal fluorescence by lipofuscin).
Clinical Presentation
Bilateral, symmetrical, painless central‐vision loss.
Early stages may appear ophthalmoscopically normal → mislabelled as non-organic/"malingering".
Visual acuity: slow decline to , then faster drop to or worse (often by age 20).
Macular atrophy (indication of disease progression)
Fundus Findings
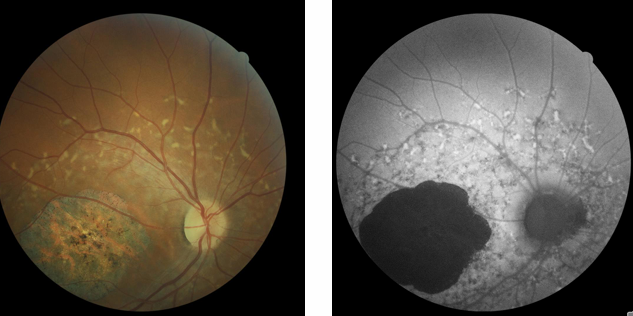
Macular ‘beaten bronze’ atrophy of RPE.
Numerous yellow, pisciform (fish-tail) flecks spread over posterior pole → term Fundus flavimaculatus (FF).
Ancillary Tests
ERG: normal early; later may show cone dysfunction.
Fluorescein angiography (FA): central hyperfluorescent window defect + dark choroid. glowing background
silent choroid - surrounding region of hyperfluorescence.
Due to lipofuscin pigment blocking fluorescence of choroidal vessels.
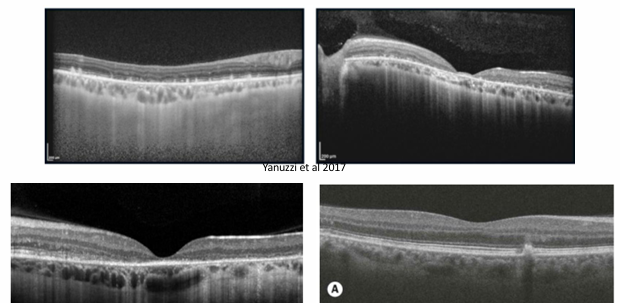
OCT: outer-retinal thinning, loss of ellipsoid zone, hyper-reflective flecks.
Fundus autofluorescence (FAF): Lipofuscin pigments in the retina exhibit auto fluorescent propertie
Flecks = hyper-autofluorescent.
Areas of macular atrophy = hypo-autofluorescent.
Management / Prognosis
No proven disease-modifying therapy.
Avoid high-dose vitamin A (may accelerate lipofuscin accumulation).
Low vision services and support services
Genetic counselling
Gene therapy and stem cell trials have been initiated and show promising results as potential treatments in the future.
Best Vitelliform (Best Disease)
uncommon hereditary, usually bilateral macular dystrophy
mild vision loss, slow progression
Genetics / Epidemiology
Autosomal dominant (mutation in VMD2/Bestrophin-1 on chromosome 11).
Penetrance high but variable expressivity; family history almost always positive.
Pathophysiology
Mutant bestrophin-1 alters RPE chloride conductance → impaired phagocytosis & lipofuscin build-up.
Classic Fundus Appearance
Yellow material (lipofuscin) accumulates at the macula
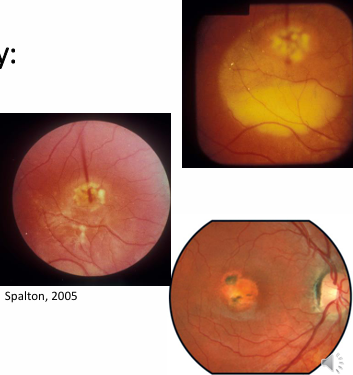
Solitary, well-circumscribed yellow “egg-yolk” lesion (1–2 disc diameters) at the fovea.
Sometimes extra-foveal satellite lesions.
Dark area on Fluorescein angiography
Lesion is intensely hyper-autofluorescent; dark on FA due to blockage.
Electrodiagnostics
Electro-oculogram (EOG) markedly abnormal (Arden ratio <).
ERG usually normal → helps distinguish from other macular dystrophies.
Stages (natural history)
Pre-vitelliform: normal fundus, abnormal EOG.
Vitelliform: classic “egg-yolk” lesion at macula.
Pseudohypopyon: Yellowish material may gravitate inferiorly.
Vitelli-ruptive: lesion breaks into “scrambled-egg”; acuity reduces.
Atrophic / cicatricial: appears as mottled RPE pigment and an oval area of atrophy
sub retinal neovascularisation can develop
OCT Correlates
Hyper-reflective dome between RPE & photoreceptor layer (vitelliform material).
Disruption of ellipsoid zone as disease advances.
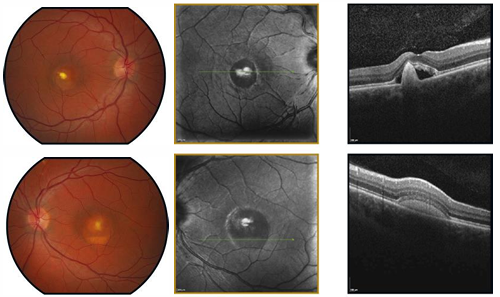
Course / Management
Onset in early childhood; many (over 75%) maintain in one eye until >50 yrs.
strong family history
Vision loss after CNV, scarring, or geographic atrophy ≥ middle age.
No treatment available
monitor with OCT/FAF
treat CNV with anti-VEGF injections.
Amsler Grid
review every 12 months
Low-vision aids; genetic counselling.
Adult-Onset Vitelliform Dystrophy (AOVD)
Form of vitelliform dystrophy that develops in adulthood
Presents in middle age or older
Consistent with normal vision, at least in the early stages, followed by often mild deterioration
Vitelliform lesions usually smaller than in Best’s disease
Regarded as one of the ‘pattern dystrophies’
A group of rare dystrophies of the RPE, characterised by accumulation of lipofuscin at the RPE (yellow, whitish, or grey deposits at the macula in a variety of morphologies).
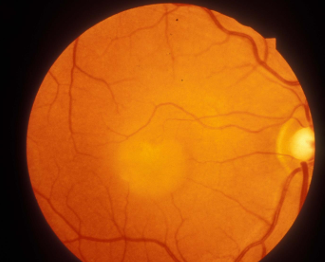
X-Linked Juvenile Retinoschisis (XLRS)
X-linked recessive → males affected, carrier females clinically silent.
Intraretinal splitting (schisis) at nerve-fiber layer & inner nuclear layer; impaired cell adhesion.
Clinical Features
Onset: first decade with unexplained central‐vision loss (~–).
Stellate or cartwheel-shaped (spokes) series of cystoid spaces at the maculae which do not leak on fluorescein angiograph
Half of all cases have a peripheral schisis as well (usually inferotemporal fundus)
ERG: “negative” pattern – markedly reduced b-wave, relatively preserved a-wave.
reduced central vision
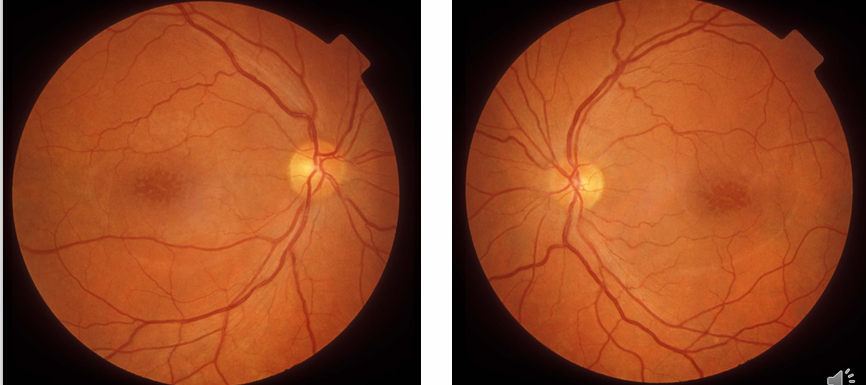
OCT
Cystic cavities within the neurosensory retina; retinal splitting plane visible.
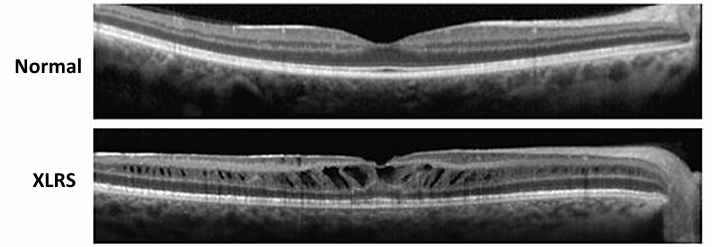
Course
Usually presents in first decade with unexplained loss of central vision (~6/9-6/12)
Vision slowly declines, reaching 6/18-6/24 by age 20
May well be 6/60 by late middle age.
If peripheral retinoschisis, can potentially be complicated by:
Vitreous haemorrhage
Retinal detachment
Management
No treatment alters the natural course of the disease.
However, topical dorzolamide and oral acetazolamide have been shown to reduce cystic spaces and foveal thickness with an increase in visual acuity.
Gene therapy is under investigation, with potential for restoring visual function in future
Low vision services may be needed.
Cone Dystrophies
Group of conditions characterised by early onset, bilateral, slowly progressive loss of central vision, due to progressive deterioration of cone photoreceptor function.
Definition & Genetics
Heterogeneous group with progressive primary cone photoreceptor dysfunction.
Inheritance: sporadic or autosomal dominant most frequent; autosomal recessive & X-linked forms exist.
Some evolve into cone-rod dystrophy when rods later affected.
Symptoms
Early blurred central vision, photophobia.
Marked colour vision loss (often red-green); frequent nystagmus.
Central/paracentral scotoma; peripheral field usually spared until late.
Signs & Imaging
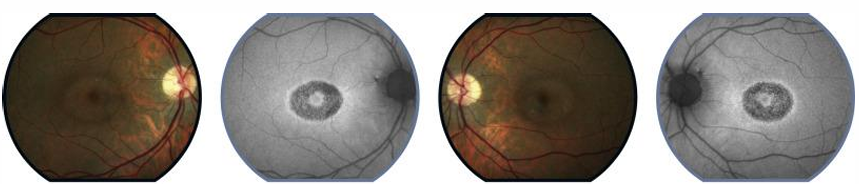
Classic ‘bull’s-eye’ maculopathy: concentric RPE atrophy with central sparing.
Fundus may exhibit non-specific central pigmentary changes at macula
central atrophic changes in later stage disease
FAF: ring of hyper- or hypo-autofluorescence outlining bull’s-eye.
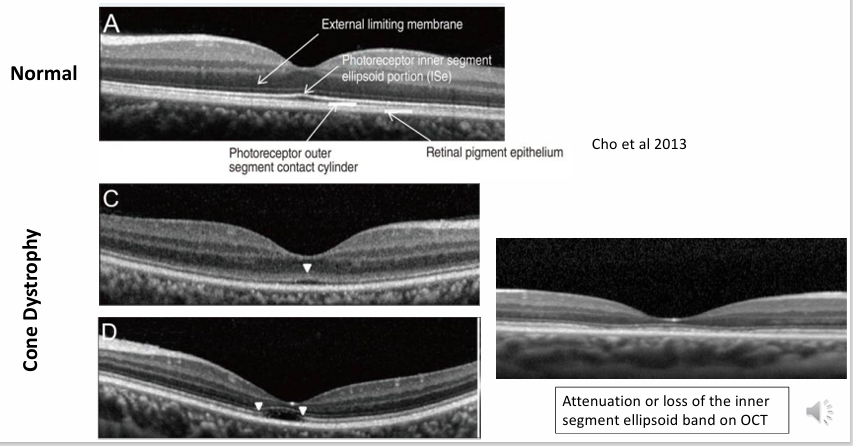
OCT: attenuation / loss of inner segment–ellipsoid (IS/ELM) band; foveal thinning.
ERG: photopic (cone) responses reduced/absent; scotopic (rod) normal early, deteriorate later if cone-rod dystrophy develops.
Course
Visual decline may start in 1st decade
VA falls to –, colour vision usually impaired.
Typically reaches (or worse) by 3rd–4th decade.
Management
No treatment currently available
Electrophysiology (ERG – abnormal cone response) important for specific diagnosis
Genetic counselling
Low vision services
Tinted spectacle to reduce glare difficulties
Cross-Topic Connections & Practical Considerations
All conditions emphasise importance of multimodal imaging (OCT, FAF, FA) + electrophysiology (ERG/EOG) for accurate phenotyping.
Lipofuscin accumulation (Stargardt, vitelliform group) is central → avoidance of vitamin A excess, exploration of pharmacological visual-cycle modulators.
Gene therapy is a recurring future avenue (ABCA4, BEST1, RS1, cone-specific genes); raises pharmacoeconomic & ethical questions regarding access, long-term surveillance, and germline editing avoidance.
Early genetic diagnosis enables informed family planning, carrier testing, and enrolment in natural-history or interventional trials.
Low-vision rehabilitation, psychosocial support, and educational accommodations remain cornerstone care across all dystrophies.
Disease | Signs | Symptoms | Associations / Inheritance | Treatment |
|---|---|---|---|---|
### Stargardt Disease (Fundus Flavimaculatus) | Macular 'beaten bronze' atrophy of RPE; numerous yellow, pisciform flecks; 'silent (dark) choroid' on FA; outer-retinal thinning, loss of ellipsoid zone, hyper-reflective flecks on OCT; flecks are hyper-autofluorescent, atrophic areas are hypo-autofluorescent on FAF. | Bilateral, symmetrical, painless central-vision loss; slow decline to , then faster drop to or worse (often by age 20). | Autosomal recessive in of cases; single most common hereditary macular dystrophy; usually manifests in childhood (<10 yrs). | No proven disease-modifying therapy; avoid high-dose vitamin A; low vision services and support; genetic counselling; gene therapy and stem cell trials promising. |
### Best Vitelliform (Best Disease) | Solitary, well-circumscribed yellow 'egg-yolk' lesion (1–2 disc diameters) at fovea (can be extra-foveal); lesion is intensely hyper-autofluorescent; dark on FA due to blockage; hyper-reflective dome between RPE & photoreceptor layer on OCT; disruption of ellipsoid zone. | Mild vision loss, slow progression. | Autosomal dominant (VMD2/Bestrophin-1 mutation on chromosome 11); high penetrance but variable expressivity. | No treatment available; monitor with OCT/FAF; treat CNV with anti-VEGF injections; Amsler Grid; review every 12 months; low-vision aids; genetic counselling. |
### Adult-Onset Vitelliform Dystrophy (AOVD) | Vitelliform lesions, usually smaller than in Best's disease; yellow, whitish, or grey lipofuscin deposits at macula. | Consistent with normal vision early, followed by often mild deterioration. | Develops in adulthood; regarded as one of the 'pattern dystrophies'. | Not explicitly stated in note, likely similar to Best's for monitoring. |
### X-Linked Juvenile Retinoschisis (XLRS) | Stellate or cartwheel-shaped (spokes) series of cystoid spaces at maculae (do not leak on FA); cystic cavities within neurosensory retina, retinal splitting plane visible on OCT; peripheral schisis (usually inferotemporal fundus) in half of cases. | Unexplained central-vision loss (–) in first decade; vision slowly declines, reaching – by age 20, may be by late middle age. | X-linked recessive (males affected, carrier females clinically silent). | No treatment alters natural course; topical dorzolamide and oral acetazolamide may reduce cystic spaces and increase VA; gene therapy under investigation; low vision services. |
### Cone Dystrophies | Classic 'bull's-eye' maculopathy (concentric RPE atrophy with central sparing); non-specific central pigmentary changes at macula, central atrophic changes later; FAF shows ring of hyper- or hypo-autofluorescence; OCT shows attenuation/loss of inner segment–ellipsoid (IS/ELM) band, foveal thinning. | Early blurred central vision, photophobia; marked colour vision loss (often red-green); frequent nystagmus; central/paracentral scotoma; peripheral field usually spared until late. | Heterogeneous group, sporadic or autosomal dominant most frequent; autosomal recessive & X-linked forms exist; some evolve into cone-rod dystrophy. | No treatment currently available; electrophysiology (ERG) important for diagnosis; genetic counselling; low vision services (tinted spectacles). |