Chapter 11 - Molecular Structure
- Covalent bond - An electron pair shared between two neighboring atoms.
- Ionic bond - Here the cohesion arises from the Coulombic attraction between ions of opposite charge.
- Major approaches to the calculation of the molecular structure * Valence bond theory * Molecular orbital theory
The Born-Oppenheimer approximation
- Born-Oppenheimer approximation - It is supposed that the nuclei, being so much heavier than an electron, move relatively slowly and may be treated as stationary while the electrons move in their field.
- Molecular potential energy curve - Obtained when more than one molecular parameter is changed in a polyatomic molecule.
- Equilibrium bond length (Re) - The internuclear separation at the minimum of the curve.
- The bond dissociation energy (Do) - It’s closely related to the depth of the minimum below the energy of the infinitely widely separated and stationary atoms.
Valence-bond theory
- Valence bond theory - The first quantum mechanical theory of bonding to be developed.
11.1 Homonuclear diatomic molecules
- Bond in valence bond theory - A bond is regarded as forming when an electron in an atomic orbital on one atom pairs its spin with that of an electron in an atomic orbital on another atom.
- σ bond (sigma bond) - The electron distribution described by the wavefunction of this equation. It has a cylindrical symmetry around the internuclear axis. It’s called this because, when viewed along the internuclear axis, it resembles a pair of electrons in an s orbital.
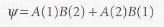
- π bond - It arises from the spin pairing of electrons in two p orbitals that approach side-by-side. It is so called because viewed along the internuclear axis, a π bond resembles a pair of electrons in a p orbital.
11.2 Polyatomic molecules
- Promotion - The excitation of an electron to an orbital of higher energy.
Molecular orbital theory
- Homonuclear diatomic molecules - Formed from two atoms of the same element.
- Heteronuclear diatomic molecules - Diatomic molecules formed from atoms of two different elements.
11.3 The hydrogen molecule-ion
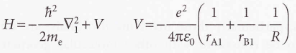
Linear combinations of atomic orbitals
- Linear combination of atomic orbitals (LCAO) - An approximate molecular orbital formed from a linear combination of atomic orbitals.
Bonding orbitals
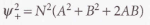
The total probability density is proportional to the sum of →
- A^2 - The probability density if the electron were confined to the atomic orbital A.
- B^2 - The probability density if the electron were confined to the atomic orbital B.
- 2AB - Extra contribution to the density. * Overlap density - It represents an enhancement of the probability of finding an electron in the internuclear region.
- Bonding orbital - An orbital which, if occupied, helps to bind two atoms together.
- σ electron - An electron that occupies a σ orbital.
Antibonding orbitals
- Antibonding orbital - An orbital that, if occupied, contributes to a reduction in the cohesion between two atoms and helps to raise the energy of the molecule relative to the separated atoms.
- Inversion symmetry - The behavior of the wavefunction when it is inverted through the center of the molecule.
- Gerade symmetry - An identical value of the wavefunction.
- Ungerade symmetry (Odd symmetry) - Same size but opposite sign of the wavefunction.
Photoelectron spectroscopy
- Photoelectron spectroscopy (PES) - It measures the ionization energies of molecules when electrons are ejected from different orbitals by absorption of a photon of the proper energy, and uses the information to infer the energies of molecular orbitals.
- Koopmans' theorem - States that the ionization energy is equal to the orbital energy of the ejected electron.
11.4 Homonuclear diatomic molecules
- Polar bond - A covalent bond in which the electron pair is shared unequally by the two atoms.
- Variation principle - If an arbitrary wavefunction is used to calculate the energy, the value calculated is never less than the true energy.
- Trial wavefunction - The arbitrary wavefunction.
11.5 Heteronuclear diatomic molecules
Molecular orbitals for polyatomic systems
11.6 The Huckel approximation
Huckel approximations
- All overlap integrals are set equal to zero.
- All resonance integrals between non-neighbors are set equal to zero.
- All remaining resonance integrals are set equal (to β).
11.7 Computational chemistry
- Hartree-Fock equations
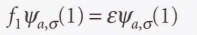 * The Fock operator (f1)
* The Fock operator (f1)
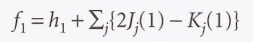 * The Coulomb operator (J)
* The Coulomb operator (J)
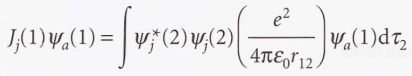 * The exchange operator (K)
* The exchange operator (K)
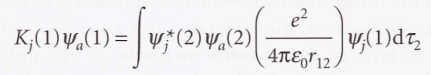
- Roothaan equations
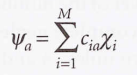 * F → The matrix formed from the Fock operator
* F → The matrix formed from the Fock operator
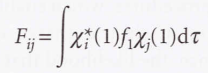 * S → The matrix of overlap integrals
* S → The matrix of overlap integrals
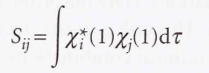
Semi-empirical and ab initio methods
- Semi-empirical methods - Where many of the integrals are estimated by appealing to spectroscopic data or physical properties such as ionization energies, and using a series of rules to set certain integrals equal to zero.
- Ab initio methods - Here an attempt is made to calculate all the integrals that appear in the Fock and overlap matrices.
- The Fock matrix
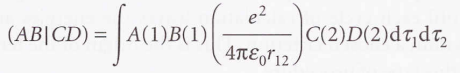
- Complete neglect of differential overlap (CNDO) - In which all integrals are set to zero unless A and B are the same orbitals centred on the same nucleus, and likewise for C and D.
- Kohn-Sham equations - They are found by applying the variation principle to the electron energy.
\
11.8 The prediction of molecular properties
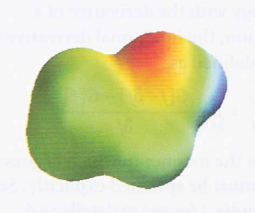
Electron density and the electrostatic potential surfaces
- Isodensity surface - A surface of constant total electron density.
- Solvent-accessible surface - Here the shape represents the shape of the molecule by imagining a sphere representing a solvent molecule rolling across the surface and plotting the locations of the center of that sphere.
- Electrostatic potential surface (An 'elpot surface') - In which net positive charge is shown in one color and net negative charge is shown in another, with intermediate gradations of color.
\
\