Autosomal Dominant Disease - 26.01.26
Medieval Impairments Disorders Overview
Focus on common genetic inheritance patterns, specifically autosomal dominant disorders.
Autosomal Dominant Disorders
Occur due to mutations in genes located on autosomes (not X or Y chromosomes).
Dominant Inheritance: Disorder manifests with just one copy of the mutated allele in the heterozygous state.
Key Characteristics of Autosomal Dominant Inheritance
Generational Presence: Disorders appear in every generation of a pedigree due to the dominant nature.
Example: If a female parent carries an autosomal dominant condition, all her children may inherit it.
Equal Sex Distribution: Males and females are equally affected, unlike X-linked disorders.
Probability of Inheritance: There is a 50% chance an affected parent will pass the disorder to their offspring.
Pedigree Analysis
Understanding pedigrees is critical for examination purposes. Pedigrees help identify how traits are passed through generations.
Important to note that specific characteristics can help differentiate inheritance patterns:
Every generation affected = likely autosomal dominant.
Equal distribution of affected males and females.
Key Terms
Penetrance: The percentage of individuals with a pathogenic variant who actually manifest the disorder.
Expression: Variability in how a genetic disorder presents physically among individuals carrying the mutation.
Examples of Autosomal Dominant Disorders
1. Familial Hypercholesterolemia
Characterized by high cholesterol levels leading to early coronary artery disease.
Caused by heterozygous pathogenic variant of the LDL receptor gene.
Symptoms include lipid deposits in skin and increased cholesterol in the blood due to reduced LDL receptor function.
Mechanism: Loss of 50% LDL receptor function leads to impaired cholesterol clearance. Treatment includes diet and statins.
Pathway Complexity
The dysfunction of the LDL receptor may arise from various mutations affecting:
Receptor synthesis.
Transport to Golgi apparatus.
Binding capability to cholesterol.
2. Neurofibromatosis Type 1
Characterized by benign tumors, café au lait spots.
Caused by pathogenic variants in the NF1 gene (a tumor suppressor).
Leads to unrestrained cellular growth, manifesting as tumors.
Full penetrance with variability in the expression of symptoms.
3. Marfan Syndrome
Symptoms: Tall stature, elongated limbs, dislocated lenses, and aortic dilation.
Caused by pathogenic variants in the fibrillin-1 gene.
Loss of structural support leads to connective tissue issues.
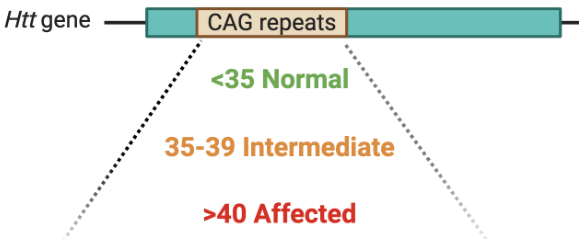
4. Huntington’s Disease
A progressive neurological disorder with a gain of function, typically expressed later in life (age-dependent expression).
100% penetrance by age 80.
Caused by CAG repeat expansions in the HTT gene.
More repeats lead to earlier onset and greater severity of symptoms.
5. Familial Adenomatous Polyposis
Fully penetrant disorder caused by mutations in the APC gene.
Manifested by numerous benign colon polyps and high cancer risk if untreated.
6. Achondroplasia
Characterized by short stature and disproportionate limb lengths due to a mutation in the FGFR3 gene.
Not always familial; new mutations can happen, leading to affected children from unaffected parents.
Fully penetrant disorder where offspring have a 50% chance of being similarly affected.
7. Charcot-Marie-Tooth Disease Type 1A (CMT1A)
Genetic Mutation: Primarily caused by a 1.51.5 Mb duplication on chromosome 17p11.217p11.2, which includes the PMP22 gene.
Effects: The duplication leads to increased PMP22 protein dosage (dosage effect), causing abnormal myelin sheath formation and maintenance in the peripheral nervous system. This results in slowed nerve conduction.
Progression: A slowly progressive peripheral neuropathy. Symptoms usually begin in the first or second decade of life with distal muscle weakness (e.g., foot drop), muscle atrophy in hands and feet, and sensory loss.
8. Osteogenesis Imperfecta (Type I)
Genetic Mutation: Pathogenic variants in the COL1A1 or COL1A2 genes, which encode the alpha chains of Type I collagen.
Effects: Mutations result either in a reduced quantity of normal collagen or the production of abnormal collagen (dominant negative effect), leading to weakened connective tissue and bone matrix.
Progression: Characterized by multiple fractures often occurring with minimal trauma. While fractures occur frequently during childhood, the rate often decreases after puberty. Secondary complications include blue sclerae, hearing loss, and dental issues (dentinogenesis imperfecta).
Mechanisms of Autosomal Dominant Diseases
Haploinsufficiency: Loss of gene dosage leads to reduced functional protein amounts insufficient for normal function.
Dominant Negative Variants: Pathogenic variant interferes with the function of the normal allele.
Gain of Function Variants: Mutations cause increased activity or new function that disrupts normal physiological processes.
Examples of Mechanism Functions
Haploinsufficiency: Seen in conditions like Familial Hypercholesterolemia where insufficient LDL receptors impair cholesterol clearance.
Dominant Negative Variants: Marfan syndrome involves dysfunctional fibrillin-1 polymers, where even a few defective proteins disrupt the entire structure.
Gain of Function: Huntington’s disease where CAG repeats cause protein aggregates leading to neuronal degeneration.
Conclusion
Understanding autosomal dominant inheritance is crucial.
Students must be familiar with identifying conditions from symptoms and understanding their genetic basis for examinations.
Mechanisms of pathogenic variants are also important for grasping the implications of these disorders in a clinical context.