Chapter 16
16.1 Electrophilic Aromatic Substitution Reactions: Bromination
In bromination, a bromine atom replaces a hydrogen on benzene. But Br₂ by itself is not electrophilic enough for benzene to attack well. So we use a Lewis acid catalyst, usually FeBr₃. The lecture says FeBr₃ is a common Lewis acid catalyst for bromination and that it activates the electrophile, making it more electrophilic.
Here is the mechanism in easy words.
First, Br₂ reacts with FeBr₃. The Lewis acid pulls electron density away from Br₂, making one bromine much more positive in character. That is the activated electrophile.
Second, one of the benzene π bonds attacks that electrophilic bromine. At this moment, benzene stops being aromatic, and the arenium ion forms. This is the slow step and the most important step in deciding reaction rate.
Third, FeBr₄⁻ or another base removes the proton from the carbon that just got bromine. The electrons from that C–H bond go back into the ring and restore aromaticity. Now you get bromobenzene plus HBr, and FeBr₃ is regenerated.
Why is this harder than addition to an alkene? Because benzene is more stable than a normal alkene, so the activation energy to form the arenium ion is higher. The slides explicitly compare benzene and alkenes and say benzene has a larger activation energy because aromatic stabilization is lost in the non-aromatic intermediate.
What you need to remember for bromination is very simple. The reagents are Br₂ and FeBr₃. The product is bromobenzene. The mechanism is activation of Br₂, attack by the ring, arenium ion, deprotonation, aromaticity restored.
Section 16.1 check-your-understanding questions
Question 1: Why does benzene do substitution instead of addition?
Answer: Because addition would permanently destroy aromaticity, while substitution allows aromaticity to be restored at the end.
Question 2: What is the role of FeBr₃?
Answer: It is a Lewis acid catalyst that activates Br₂ and makes the bromine more electrophilic.
Question 3: What is the arenium ion?
Answer: It is the non-aromatic carbocation intermediate formed after the ring attacks the electrophile.
Question 4: What are the two steps common to all EAS reactions?
Answer: Addition of the electrophile, then elimination of H⁺ to restore aromaticity.
16.2 Other Aromatic Substitutions
This section is basically “same EAS idea, different electrophile.”
Fluorination
The slides say fluorine is not usually added to aromatic rings by a standard EAS method. One method uses Selectfluor with strong acid, TfOH, as a source of electrophilic fluorine.
For your course, this is more of a recognition point than a big mechanism point. Just remember that aromatic fluorination is not usually done with plain F₂ the way bromination uses Br₂.
Chlorination
Chlorination works just like bromination. The lecture says the usual reagents are Cl₂ and FeCl₃. So the mechanism is the same pattern: activate chlorine, ring attacks, arenium ion, deprotonation, aromaticity returns.
Iodination
I₂ is less reactive toward aromatic rings, so an oxidant such as H₂O₂ or CuCl₂ is added. The slides say this forms an I⁺-like species that reacts with the ring.
So the big difference is that iodine needs help generating a strong enough electrophile.
Nitration
This is one of the most important reactions in the chapter.
The reagents are concentrated HNO₃ and concentrated H₂SO₄. The actual electrophile is not neutral nitric acid. The real electrophile is the nitronium ion, NO₂⁺. The lecture clearly states that the active electrophile is NO₂⁺, generated by dehydration of HNO₃ with H₂SO₄.
Mechanism in easy words:
First, sulfuric acid protonates nitric acid.
Then water leaves, giving NO₂⁺, the nitronium ion.
Then benzene attacks NO₂⁺ and forms the arenium ion.
Finally, a base removes the proton and aromaticity returns, giving nitrobenzene.
This reaction is very important because the nitro group can later be changed into an amine. The lecture shows that aromatic nitro groups can be reduced to aromatic amines using Fe, Zn, or Sn in acid, followed by basic workup to deprotonate the ammonium product.
That means a very common synthetic trick is:
benzene → nitration → nitrobenzene → reduction → aniline.
Sulfonation
Sulfonation adds SO₃H to the aromatic ring. The slides say the active electrophile is SO₃H⁺, generated from H₂SO₄ and SO₃.
Mechanistically it is still EAS. The ring attacks the sulfur electrophile, forms an arenium ion, then loses a proton to restore aromaticity.
The really important special feature is that sulfonation is reversible. The lecture says strong acid favors sulfonation, while high temperature and dilute aqueous acid favor desulfonation.
This becomes very useful in synthesis. You can temporarily put SO₃H on a ring as a blocking group, do another reaction, then remove it later.
Section 16.2 check-your-understanding questions
Question 1: What is the real electrophile in nitration?
Answer: NO₂⁺, the nitronium ion.
Question 2: What reagents are used for chlorination?
Answer: Cl₂ and FeCl₃.
Question 3: Why is iodination harder than bromination or chlorination?
Answer: Because I₂ is not reactive enough on its own, so an oxidant is needed to generate an I⁺-like electrophile.
Question 4: What is special about sulfonation?
Answer: It is reversible. Strong acid favors adding SO₃H, and hot dilute aqueous acid favors removing it.
Question 5: How do you convert a nitro group into an amino group?
Answer: Reduce the nitro group with Fe, Zn, or Sn in acid, then do basic workup.
16.3 Alkylation and Acylation of Aromatic Rings: The Friedel–Crafts Reaction
This section is very testable because it has mechanisms and lots of limitations.
Friedel–Crafts alkylation
This reaction adds an alkyl group to benzene. The electrophile is a carbocation-like species generated from an alkyl halide and AlCl₃. The slides state that Friedel–Crafts alkylation introduces an alkyl group to benzene and that the electrophile is a carbocation generated from RX and AlCl₃.
Mechanism in easy words:
First, the alkyl halide interacts with AlCl₃, making the carbon attached to the halogen much more electrophilic.
Second, a carbocation or carbocation-like electrophile forms.
Third, benzene attacks that electrophile and gives the arenium ion.
Fourth, deprotonation restores aromaticity, giving alkylbenzene.
The class question on the slides asks what kind of alkyl halides are most suitable, based on carbocation stability. The best answer is tertiary alkyl halides, because they form the most stable carbocations. Secondary can work too. Primary are much worse and often rearrange.
Limitations of Friedel–Crafts alkylation
This is one of the most important memorization parts.
First, it only works with alkyl halides. Vinyl halides and aryl halides do not work because vinyl and aryl carbocations are too unstable. The slides say exactly that.
Second, it does not work well on strongly deactivated aromatic rings. The lecture says if a meta-directing deactivator is already on the ring, Friedel–Crafts will not occur because the carbocation electrophile is not strong enough. Examples listed include nitro and sulfonyl-type groups.
Third, amines are problematic. Even though NH₂ is usually activating in normal EAS, in Friedel–Crafts conditions the amine lone pair reacts with the Lewis acid catalyst and becomes deactivating. The lecture says this can sometimes be solved by converting the amine to an amide first.
Fourth, overalkylation can happen. Once you add one alkyl group, the ring becomes more electron-rich and reacts even faster, so it may get alkylated again. The slides say overalkylation can be avoided by using excess aromatic compound.
Fifth, carbocation rearrangements can happen, especially with primary alkyl halides. The slides specifically show hydride shifts and alkyl shifts.
That means Friedel–Crafts alkylation often gives “surprise” products if the initially formed carbocation can rearrange to a more stable one.
The worked example on the slide about 2-chloro-3-methylbutane is exactly testing that rearrangement idea.
Friedel–Crafts acylation
This is like alkylation, but instead of adding R, you add an acyl group, RCO–. Usually the reagent is an acid chloride plus AlCl₃. The slides say the active electrophile is an acylium ion and show acetyl chloride with AlCl₃ giving acetophenone.
Why is acylation often better than alkylation?
Because the acylium ion is resonance stabilized, so it does not rearrange. The lecture says no carbocation rearrangements occur during Friedel–Crafts acylation.
Also, only one acyl group is usually added. After acylation, the carbonyl-containing group is electron withdrawing, so the ring becomes less reactive toward a second EAS reaction. The slides mention that only one acyl group is added, with the reason becoming clearer in the next section.
Mechanistically, it is the same EAS pattern. First generate the acylium ion from the acid chloride and AlCl₃, then the ring attacks, then deprotonate and restore aromaticity.
Section 16.3 check-your-understanding questions
Question 1: Why are tertiary alkyl halides best for Friedel–Crafts alkylation?
Answer: Because they form the most stable carbocations.
Question 2: Why don’t vinyl halides and aryl halides work?
Answer: Because vinyl and aryl carbocations are too unstable to form under these conditions.
Question 3: Why can Friedel–Crafts alkylation give rearranged products?
Answer: Because carbocation intermediates can undergo hydride or alkyl shifts to become more stable before the ring attacks.
Question 4: Why is acylation usually cleaner than alkylation?
Answer: Because the acylium ion is resonance stabilized, so it does not rearrange, and the product is deactivated so polyacylation is not common.
Question 5: Why are amines problematic in Friedel–Crafts reactions?
Answer: Because the amine lone pair reacts with the Lewis acid catalyst and makes the ring deactivated under those conditions.
16.4 Substituent Effects in Electrophilic Substitutions
This is the heart of the chapter.
A substituent already on the ring affects two things. It affects how fast the ring reacts, and it affects where the new group goes. The lecture says existing substituents affect both reaction rate and regioselectivity.
Part 1: Activating versus deactivating
Activating means the substituted ring reacts faster than benzene. Deactivating means it reacts slower than benzene. The slides define these exactly.
Why? Because activating groups donate electron density into the ring. That stabilizes the positively charged arenium ion, lowers the activation energy, and speeds up the reaction. Deactivating groups withdraw electron density, destabilize the arenium ion, raise the activation energy, and slow the reaction. The lecture says this directly.
Part 2: Ortho/para directors versus meta directors
Ortho and para directors guide the new electrophile to the ortho and para positions. Meta directors favor the meta position. The lecture defines these directly.
The slides classify most substituents into three groups:
o/p-directing activators, o/p-directing deactivators, and m-directing deactivators.
Why groups donate or withdraw: induction and resonance
The lecture says the overall electron effect comes from induction and resonance. Induction is electron donation or withdrawal through sigma bonds because of electronegativity differences. Resonance is donation or withdrawal through overlapping p orbitals and pi systems.
Alkyl groups
Alkyl groups donate weakly by induction. They slightly increase electron density in the ring, so they activate the ring. They stabilize the arenium ion formed by ortho and para attack better than the meta one, so they are ortho/para directors. The lecture specifically says alkyl substituents donate by induction and are o/p-directing.
OH, OR, NH₂, NHR, NR₂
These groups have lone pairs directly attached to the ring. They donate strongly by resonance. That makes the ring much more reactive and strongly favors ortho and para substitution. The lecture says hydroxyl, alkoxy, and amino substituents are strongly electron donating by resonance and are o/p-directing.
There is also a subtle point in the slides: if that heteroatom is attached to a polarized π bond, like in an amide, then donation into the ring is weaker because the lone pair is also shared with the carbonyl. This is called cross-conjugation. That is why acetanilide is less reactive than aniline.
Halogens
Halogens are the famous weird case. They are deactivating, but ortho/para directing. The lecture explains why. Halogens withdraw electron density by induction, which makes the ring less reactive overall. But they can still donate by resonance with their lone pairs, and that stabilizes the ortho and para arenium ions more than the meta one. So halogens are o/p-directing deactivators.
This is one of the most common exam traps.
Strong electron-withdrawing resonance groups
Groups like NO₂, CN, CHO, COR, COOR, COOH, SO₃H and similar groups pull electron density away by resonance and induction. They make the ring less reactive, so they are deactivating, and they direct meta because the ortho and para arenium ions would place positive charge next to or on the already electron-poor region, which is especially unstable. The lecture says substituents with a positively polarized atom directly attached to the ring are meta-directing deactivators.
Quick rule to memorize
Most electron-donating groups are ortho/para activating.
Most electron-withdrawing groups are meta deactivating.
Halogens are the exception: ortho/para directing but deactivating.
The slide summary table shows exactly this pattern.
The lecture class questions here
The methoxybenzene bromination question shows anisole reacting with Br₂/FeBr₃. Methoxy is a strong ortho/para director, so the major products are the ortho and para brominated products, not the meta product. So the correct choice is A and C.
The Hammond postulate question asks what the first transition state most closely resembles. Since the first step is endergonic and produces the high-energy arenium ion, the first transition state is more product-like and resembles the arenium ion intermediate. So the correct answer is the arenium ion intermediate.
Section 16.4 check-your-understanding questions
Question 1: Is CH₃ activating or deactivating, and where does it direct?
Answer: It is activating and ortho/para directing.
Question 2: Is NO₂ activating or deactivating, and where does it direct?
Answer: It is strongly deactivating and meta directing.
Question 3: Why is OH strongly activating?
Answer: Because it donates electron density strongly by resonance through its lone pair.
Question 4: Why are halogens ortho/para directing even though they deactivate?
Answer: They withdraw by induction, which slows the reaction overall, but they donate by resonance, which stabilizes ortho and para arenium ions.
Question 5: In nitration of toluene, why is meta minor?
Answer: Because the methyl group stabilizes ortho and para arenium ions better than the meta arenium ion.
6.4 Textbook

a)Let’s look at each one.
Phenol has OH, and OH strongly donates electrons by resonance. That makes the ring much more reactive than benzene.
Toluene has CH₃, and CH₃ weakly donates electrons. So it is more reactive than benzene, but not as much as phenol.
Benzene is the standard reference.
Nitrobenzene has NO₂, and NO₂ strongly withdraws electrons. That makes the ring much less reactive than benzene.
So the order from most reactive to least reactive is:
phenol > toluene > benzene > nitrobenzene
Why?
Because:
OH strongly activates
CH₃ weakly activates
benzene is normal
NO₂ strongly deactivates
b)phenol > benzene > chlorobenzene > benzoic acid
Why?
Because:
OH strongly activates
benzene is normal
Cl deactivates
COOH strongly deactivates
c)Let’s go one by one.
Aniline has NH₂, which is a strong activator, so it reacts the fastest.
Benzene is the reference.
Bromobenzene has Br, which is deactivating, so it reacts slower than benzene.
Benzaldehyde has CHO, which is strongly electron withdrawing, so it is even less reactive.
So the order is:
aniline > benzene > bromobenzene > benzaldehyde
Why?
Because:
NH₂ strongly activates
benzene is normal
Br deactivates
CHO strongly deactivates

a) Starting compound: bromobenzene
The group already on the ring is Br.
Br is:
deactivating
but ortho/para directing
So when you nitrate bromobenzene, the incoming NO₂ goes mainly to the ortho and para positions relative to Br.
So the major products are:
o-bromonitrobenzene
which is 1-bromo-2-nitrobenzenep-bromonitrobenzene
which is 1-bromo-4-nitrobenzene
Usually the para product is often favored more because it is less crowded.
Easy way to think:
Br says: “I slow the reaction down, but if substitution happens, put the new group ortho or para to me.”
b)The group already there is NO₂.
NO₂ is:
strongly deactivating
meta directing
So bromine goes to the meta position.
The major product is:
m-bromonitrobenzene
which is 1-bromo-3-nitrobenzene
c)OH is:
strongly activating
ortho/para directing
So chlorine goes mainly to the ortho and para positions.
The major products are:
o-chlorophenol
which is 2-chlorophenolp-chlorophenol
which is 4-chlorophenol
c)The group already there is NH₂.
NH₂ is:
very strongly activating
ortho/para directing
So bromine goes to the ortho and para positions.
The expected products by directing effects are:
o-bromoaniline
which is 2-bromoanilinep-bromoaniline
which is 4-bromoaniline
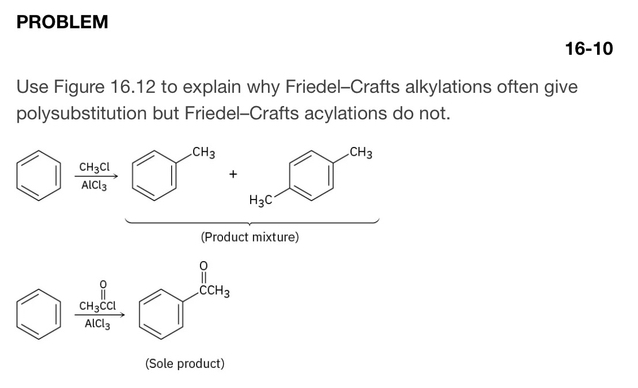
Friedel–Crafts alkylation gives polysubstitution because the alkyl group activates the ring, making it more reactive toward further substitution.
Friedel–Crafts acylation does not give polysubstitution because the acyl group deactivates the ring, making it less reactive after the first substitution.

(trifluoromethyl)benzene is less reactive than toluene toward electrophilic substitution.
This is because CF₃ strongly pulls electron density away from the ring, making it less reactive, while CH₃ donates electron density and makes the ring more reactive.

Acetanilide is less reactive than aniline because the nitrogen’s lone pair is partially tied up with the carbonyl group, so it cannot donate as much electron density to the benzene ring. As a result, the ring is less activated toward electrophilic substitution.

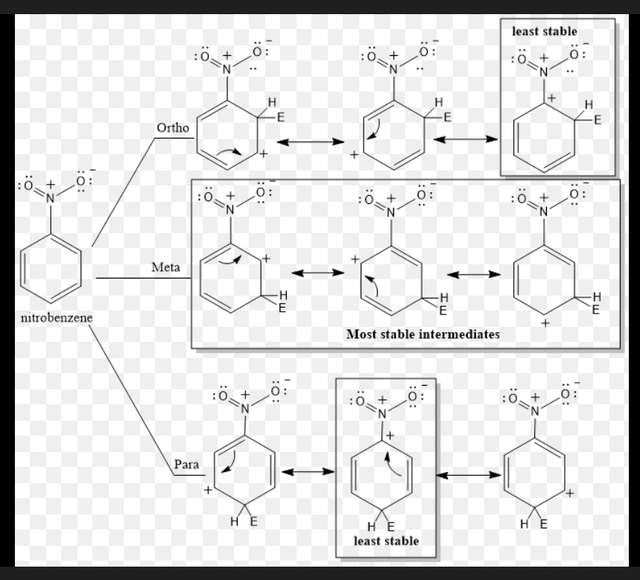
The intermediates formed from ortho and para attack are less stable because their resonance structures place a positive charge on the carbon bearing the nitro group, which is highly electron-withdrawing and destabilizes the intermediate.
The intermediate from meta attack is more stable because none of its resonance structures place the positive charge on the carbon attached to the nitro group.
16.6 Nucleophilic Aromatic Substitution, SNAr
This is the opposite flavor from EAS.
In EAS, the ring is electron-rich and attacks an electrophile.
In SNAr, the ring is electron-poor and gets attacked by a nucleophile.
The lecture says aryl halides with electron-withdrawing groups can undergo nucleophilic aromatic substitution, and that the best substrates are very electron-poor aromatic rings.
Why normal SN1 and SN2 do not happen
The slides remind you that SN1 and SN2 mechanisms do not operate on aryl substrates. An aryl carbocation is too unstable for SN1, and backside attack at an sp² carbon is blocked for SN2.
The actual SNAr mechanism
The mechanism is addition–elimination.
First, the nucleophile attacks the carbon bearing the leaving group. This breaks aromaticity and forms an anionic intermediate called the Meisenheimer complex.
Second, the leaving group departs and aromaticity is restored.
The lecture says the Meisenheimer complex has a buildup of negative charge, and electron-withdrawing groups ortho or para to the halide stabilize it by resonance. Meta EWGs do not help because they are cross-conjugated.
This means the best SNAr substrates are things like nitro-substituted chlorobenzenes where the nitro groups are ortho and/or para to the leaving group.
The comparison slide is very helpful. It says EAS is favored by electron-donating groups and has a cationic arenium ion intermediate, while SNAr is favored by electron-withdrawing groups and has an anionic Meisenheimer complex intermediate. It also says that in EAS, EWGs are meta directing, but in SNAr, EWGs are ortho/para directing because they stabilize the negative-charge intermediate.
That contrast is a very nice exam point.
Section 16.6 check-your-understanding questions
Question 1: Why do nitro groups help SNAr?
Answer: Because they withdraw electron density and stabilize the negatively charged Meisenheimer complex by resonance.
Question 2: Why don’t SN1 and SN2 usually happen on aryl halides?
Answer: SN1 would require an unstable aryl carbocation, and SN2 backside attack at an sp² carbon is not favorable.
Question 3: Which positions of EWGs help SNAr the most?
Answer: Ortho and para to the leaving group. Meta does not help much.
16.7 Benzyne
Sometimes aryl halides without electron-withdrawing groups still react with strong base, but only under very harsh conditions.
The lecture says halobenzenes lacking EWGs do not normally react with nucleophiles under normal conditions, but under forcing conditions chlorobenzene can give phenol with dilute NaOH at 340 °C and 170 atm.
That reaction does not use the normal SNAr mechanism. Instead, it goes through benzyne.
Evidence for benzyne
The isotopic labeling experiment shown in the slides is very important. When bromobenzene labeled at one carbon reacts and gives aniline, the label ends up 50:50 at two adjacent positions. That means a symmetrical intermediate formed first. The lecture says this strongly implies formation of a symmetrical product intermediate.
Mechanism in easy words
A very strong base removes a proton adjacent to the halogen.
Then the halide leaves.
That creates benzyne, which is a highly reactive intermediate that looks like benzene with a “triple bond” in the ring.
Then the nucleophile adds to one end or the other of that benzyne bond.
Finally, protonation gives the substituted product.
Because benzyne is symmetrical, attack can happen at either end, so mixtures can result. The slides say benzyne is the intermediate formed by elimination of HX.
Structure of benzyne
The lecture says benzyne contains a highly distorted alkyne. The carbons remain sp² hybridized, and the second π bond is weak because it comes from overlap of sp² orbitals instead of normal p–p overlap.
You do not need to overcomplicate this. Just remember benzyne is very strained and very reactive.
Section 16.7 check-your-understanding questions
Question 1: Under what kind of conditions does benzyne chemistry happen?
Answer: Very harsh, forcing conditions with strong base and high temperature/pressure.
Question 2: Why do you often get mixtures from benzyne intermediates?
Answer: Because benzyne is symmetrical, so nucleophiles can add at either end.
Question 3: Is benzyne the same as a normal alkyne?
Answer: No. It is highly distorted and much weaker/more reactive than a normal alkyne.
16.8 Oxidation of Aromatic Compounds
This section is mostly about the benzylic position.
The lecture says benzene rings themselves are inert to strong oxidants like KMnO₄ because of aromatic stabilization, but benzylic side chains can be oxidized to benzoic acid. It also says that at least one benzylic hydrogen is necessary, or else no reaction occurs.
What is the benzylic position?
It is the carbon directly attached to the aromatic ring.
So in ethylbenzene, the CH₂ next to the ring is benzylic.
In tert-butylbenzene, the carbon attached to the ring has no hydrogens, so there is no benzylic hydrogen.
The key oxidation rule
No matter how long the alkyl side chain is, if there is at least one benzylic hydrogen, strong oxidation usually turns the whole side chain into COOH.
So:
toluene becomes benzoic acid.
ethylbenzene becomes benzoic acid.
propylbenzene becomes benzoic acid.
But tert-butylbenzene does not react because there is no benzylic H. The lecture explicitly shows this idea.
The oxidation class question is testing exactly that: only side chains with benzylic hydrogens get oxidized.
The slides also mention NBS with radical initiator for benzylic bromination. That is related because the benzylic position is especially reactive in radical chemistry too.
Section 16.8 check-your-understanding questions
Question 1: What does KMnO₄ do to an alkylbenzene with a benzylic hydrogen?
Answer: It oxidizes the benzylic side chain all the way to a carboxylic acid.
Question 2: Will tert-butylbenzene oxidize to benzoic acid?
Answer: No, because it has no benzylic hydrogens.
Question 3: Does the aromatic ring itself usually get oxidized by KMnO₄?
Answer: No, the ring is resistant because of aromatic stabilization.