Ageing
Defining Ageing
Chronological age Functional age
Primary aging
Decline in biological function - affects us all
Occurs in the context of overall good health i.e. “normal” ageing process
Secondary ageing
Decline in function die to hereditary defects and negative environmental influences
disease, poor lifestyle choices, environmental pollution and psychological stress
Biological Ageing
Biological ageing or senescence = impact of time on the body
Changes range from those affecting its cells and their function to those affecting the whole organism
Linked to shortening of telomeres
Telomeres are repeated sequences of nucleotides on DNA
6 base pairs, TTAGGG on one strand bound to AATCCC on the other strand (repeated many times)
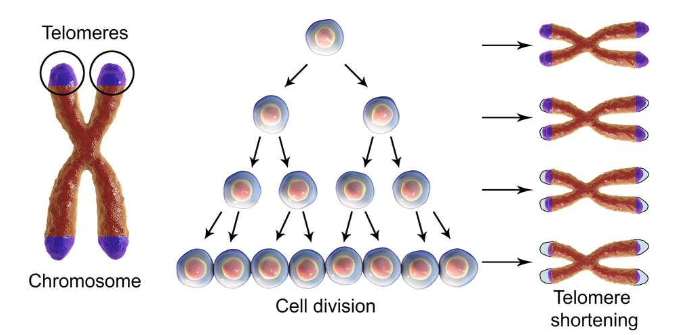
Cells age based on the number of times they have replicated
More damage to your cells the more your cells need to replicate (i.e. tumours/cancers)
Cells may self-destruct if the damage cannot be easily repaired
Normal diploid cell can replicate
~50 times before genetic material no longer able to be copied
Neurons are different; don’t replicate – have to be protected from damage (by glial cells)
Neurodegeneration
The gradual loss of neural structure or function
Neurodegenerative conditions → chronic and progressive loss
Many things kill neurons
Oxidative stress
Lifestyle
Neuroinflammation
Glutamate
Many proteins can have toxic effects
Prions (misfolded proteins)
Beta-amyloid (plaques)
Hyperphosphorylated tau (tangles)
Alpha-synuclein (Lewy bodies)
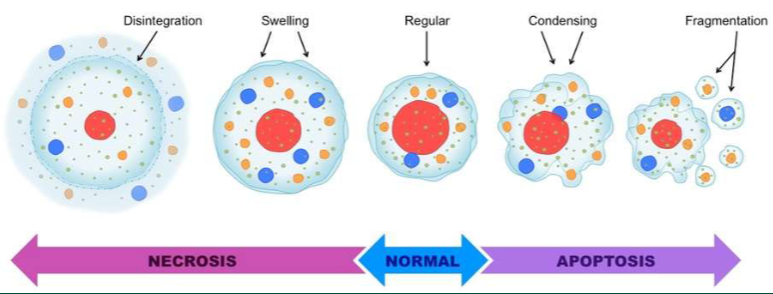
Neurons may die as a result of
Necrosis
toxins, products of metabolism, cause direct damage to the cells
Apoptosis
programmed cell death, may be initiated due to presence of certain agents
One of main systems involved in cellular aging is the cholinergic system (NT - acetylcholine: ACh) (See Cholinergic system overview in additional materials). (Lee & kim., 2022)
Cholinergic neurons in the basal forebrain = 1st cells to be affected in degenerative disorders, followed by other subcortical regions
e.g. PD, Down-syndrome, progressive supranuclear palsy, CJD,
Korsakoff's syndrome, TBI. (Schliebs & Arendt, 2011)
loss of ACh in the hippocampus contributes to memory decline with ageing
Cortisol (stress) interferes with hippocampal morphology, suppresses proliferation and reduces volume (Kim, Pellman & Kim, 2015)
Autonomic nervous system changes
Increases in blood pressure
Increases in cortisol release
More inflammation responses in the peripheral areas.
Weakening muscular strength (neuromuscular junction changes)
Sympathetic nerve activation decreases
Autonomic symptoms in older adults explains about 20% of Health-Related Quality of Life measures and 32% of depressive mood states. (Renno-Busch et al., 2021. Front. Neurol.)
Structural changes
Sulci and gyri become more pronounced
Ventricle space increases
Grey and white matter reductions (cell death or myelin destruction or integrity reduction).
Lifetime loss approx. 100grams or 7%
Reductions efficiency
Degradation in Grey and white matter* shows that those that demonstrate poor cognitive and/or motor functions show largest degradation
* measured via diffusor tensor imaging via DTI and FA techniques
Alzheimer’s Disease (AD) Dementia
Slow & progressive disease of the brain
Initially manifests as impairment of memory
Leading to problems with reasoning, planning, language, perception & emotion (e.g. depression)
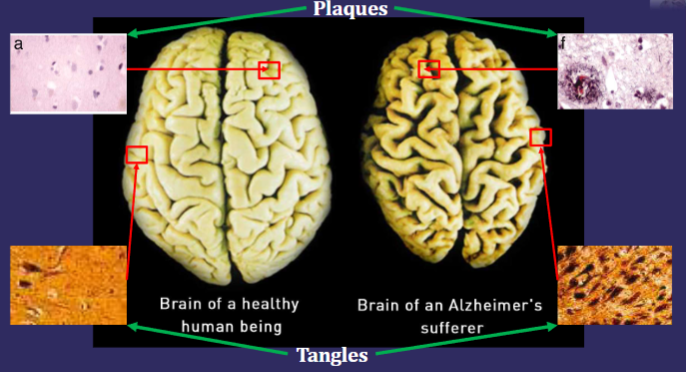
Characterized by cerebral atrophy
i.e., loss of brain cells and important brain systems like the cholinergic system
At autopsy, AD brains contain:
Neurofibrillary tangles (intracellular)
Senile plaques (extracellular)
Demographics:
Increasing age = increasing prevalence of AD
≈50% of people aged 85+ have disease
4.4% % of those with AD were age 65+ yrs in Europe,
7.7% over 70yrs have AD
3.9% worldwide prevalence when 60+ yrs old
Declines in death rates after age 65 mean that more people will survive to the oldest ages, where risk of AD is greatest.
Diagnosis:
No certainty till post-mortem when brain tissue can be examined
During life, a patient can be diagnosed with “probable AD”
Neuroimaging (e.g., CT Scan, MRI, FDG-PET)
Medical and psychiatric history
Physical examination
Psychometrics/cognitive function assessments
Mini mental state exam (MMSE)
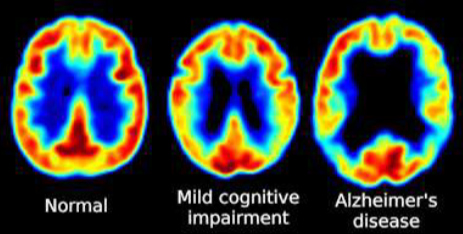
Stages:
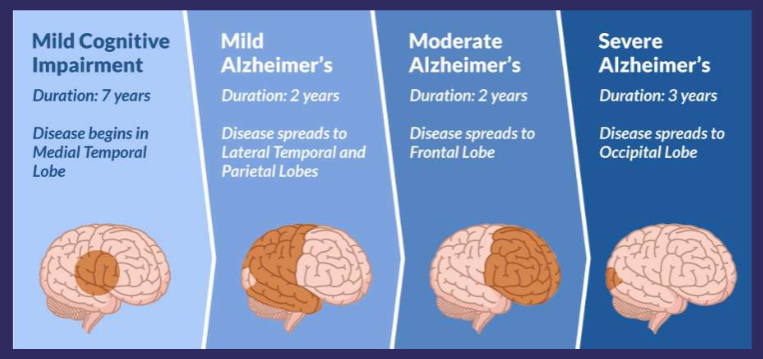
Is AD typical of old age?
Amyloid Cascade hypothesis
Plaques composed of a protein called beta- amyloid (Abeta, Aβ), extremely toxic to brain cells in high levels
Failure in the metabolism of Amyloid precursor protein (APP) leads to the formation of Aβ and their aggregation as senile plaques
Amyloid precursor protein (APP)
Integral membrane glycoprotein expressed in many tissues
Concentrated in the synapses of neurons
Thought to have a role in synapse formation and neural plasticity
Precursor protein of Aβ peptide
Proteolysis (degradation of proteins by protease enzymes) of APP produces Aβ
Synthesized by the cleavage of APP by enzymes
β-secretase and γ-secretase
Aβ Peptide
The Aβ peptide is hydrophobic and self-aggregating
Aβ accumulates to form amyloid plaques (senile plaques)
Plaques interfere with neurotransmission e.g. Russell et al., (2012)
Aβ encourages tau proteins to form tangles inside neurons, although the mechanism is unclear
Aβ also directly toxic: dimers cause damage at synaptic clefts
Strategies to decrease the production of Aβ, stimulate the clearance of Aβ formed or prevent the aggregation of Aβ into amyloid plaques are being pursue
Tau and NFTs
In AD, tau undergoes hyperphosphorylation causing microtubules to collapse
Tau proteins clump together to form neurofibrillary tangles (NFTs)
AD is a true tauopathy
Genetics - ApoE
ApoE (Apolipoprotein E) gene
ApoE has a role in cellular repair and carrying cholesterol
Three variants:
E2 – 7%, linked to atherosclerosis & Parkinson’s
E3 – 79%, “neutral” type
E4 – 14%, linked to AD and other unfavourable outcomes
E4 variant is largest genetic risk factor for late- onset sporadic AD
ApoE enhances proteolytic break-down of Aβ
ApoE-ε4 is not as effective at catalyzing these reactions
Genetics
The accumulation of Aβ1–42 is dependent upon the cleavage of the β-secretase and the γ-secretase enzymes
Presenilin genes (PS1 and PS2) are involved in γ- secretase enzyme activity, and mutations in presenilins often lead to Aβ1-42 accumulation as found in AD patients
Transgenic mouse models used to study potential gene therapy for AD – “knockout” mice that inhibit γ-secretase
But mice showed reduced Aβ - still showed neurodegeneration suggesting that γ-secretase does have important normal function
Treating AD
No cure
Preventative actions
Developing treatments:
Memantine
Acts on glutamatergic system
Blocks NMDA-type glutamate receptors
Reduces excitotoxicity
Brand names:
Axura, Akatinol, Abixa, Memox Namenda
Modest effect in moderate-to-severe AD
Cholinesterase inhibitors
Significant treatment effects
Consistently better than placebo
Disease eventually continues to progress
Average effect size is modest
Global changes in cognition, behaviour, and functioning have been detected by both physicians and caregivers
Critical targets for the effective management of AD by an increase in the availability of acetylcholine in the brain regions and decrease in the Ab deposition
Potential protective agents
Higher education (Cognitive reserve)
Ongoing intellectual stimulation
Brain training games e.g. Lumosity, Nintendo DS, not yet proven to be effective (Owen et al., 2010)
Physical and leisure/social activities
Physical exercise (in Tg mice) inhibits levels of Aβ ≈ benefit the brain by making it more resistant to stress- induced neuron cell damage (Um et al., 2008)