cloning - recombinant DNA
Les principes de la technique
Nécessité d’analyser un fragment d’ADN spécifique ou de recombiner différents fragments d’ADN
Les fragments sont coupés, isolés et amplifiés afin d’obtenir des millions de copies
La méthode repose la capacité des bactéries à se reproduire rapidement et à recopier un fragment de DNA des million de fois à clonage
Après amplification dans les bactéries, les fragments peuvent être analysés plus facilement ou utiliser àd’autres fins
Interêt:
Production de protéines recombinantes :
But: Permettre la production de grandes quantités d'une protéine spécifique, souvent à des fins thérapeutiques ou industrielles.
Comment: Un gène codant pour la protéine d'intérêt est inséré dans un vecteur (plasmide, virus...), qui est ensuite introduit dans une cellule hôte (bactérie, levure, cellule animale...). La cellule hôte, en se multipliant, produit également la protéine recombinante.
Exemples: Production d'insuline, d'hormone de croissance, d'anticorps thérapeutiques, d'enzymes industrielles...
Étude de la fonction des gènes :
But: Comprendre le rôle d'un gène spécifique dans un organisme.
Comment: Un gène est cloné et modifié (mutations, délétions...) avant d'être réintroduit dans l'organisme. L'étude des modifications phénotypiques permet de déterminer la fonction du gène.
Exemples: Identification de gènes impliqués dans des maladies, étude du développement embryonnaire, analyse de voies de signalisation..
Thérapie génique :
But: Introduire un gène fonctionnel dans des cellules pour corriger un défaut génétique responsable d'une maladie.
Comment: Un gène thérapeutique est inséré dans un vecteur (virus...), qui est ensuite utilisé pour infecter les cellules cibles. Le gène thérapeutique s'exprime dans les cellules et compense le défaut génétique.
Exemples: Traitement de certaines formes de myopathie, de mucoviscidose, de maladies héréditaires...
Création d'organismes génétiquement modifiés (OGM) :
But: Modifier les caractéristiques d'un organisme (plantes, animaux...) à des fins agricoles, industrielles ou de recherche.
Comment: Un gène est introduit dans l'organisme, ce qui entraîne l'expression d'un nouveau caractère.
Exemples: Plantes résistantes aux herbicides ou aux insectes, animaux produisant des protéines thérapeutiques dans leur lait...
Diagnostic de maladies :
But: Détecter la présence de mutations ou d'anomalies génétiques responsables de maladies.
Comment: Des techniques de clonage et d'ADN recombinant sont utilisées pour amplifier et identifier des séquences d'ADN spécifiques associées à la maladie.
Exemples: Diagnostic de maladies infectieuses, de cancers, de maladies héréditaires...
étapes:
1. formation du vecteur plasmide avec le gène d’intérêt
ligase
il faut employer une ligase pour inseret le gène d’intérêt dans le plasmide
Elle fait la liaison covalente de l'insert d'ADN (votre gène d'intérêt) au vecteur (plasmide ou autre). Elle catalyse la formation de la liaison phosphodiester entre l'extrémité 3'-OH de l'insert et l'extrémité 5'-phosphate du vecteur, scellant ainsi les brins d'ADN et créant une molécule d'ADN continue. Pour que la ligase puisse faire son travail il faut des "cohesive ends”, compatibles.
formation des extremités compatibles
Digestion par des enzymes de restriction :
De plus on essaie d’utiliser la même enzyme pour le plasmide initial et le brin d’ADN parce que comme ça on est certain que les petits bouts cohésifs sont compatibles.
Terminal transférase :
Principe : La terminal transférase est une enzyme qui ajoute des nucléotides à l'extrémité 3' d'un brin d'ADN. Cette enzyme peut être utilisée pour ajouter des homopolymères (séquences répétées du même nucléotide) à l'extrémité 3' de deux fragments d'ADN différents. Si les deux fragments ont des homopolymères complémentaires, ils pourront s'apparier et être ligaturés.
[DESSIN]
Klenow :
Principe : La Klenow est un fragment de l'ADN polymérase I d'E. coli qui possède une activité de polymérisation et une activité exonucléasique 3' → 5'. Cette enzyme peut être utilisée pour remplir les extrémités cohésives 5' protubérantes (polymérise) ou pour transformer les extrémités cohésives 3' en extrémités franches (exonucléase).
ici on veut créer une extrémité franche, en fait ça marche aussi c’est juste moins efficace
Linkers ou adapteurs :
Principe : Les linkers sont de courts fragments d'ADN double brin qui contiennent un site de restriction. Les adapteurs sont des fragments d'ADN simple brin qui peuvent être hybridés à l'extrémité d'un fragment d'ADN. Ces outils peuvent être utilisés pour ajouter des sites de restriction ou d'autres séquences spécifiques aux extrémités des fragments d'ADN, ce qui permet de les rendre compatibles pour la ligation.
Introduction de sites de restriction aux extrémités de fragments d’ADN
linker
Choix du linker : On choisit un linker contenant le site de restriction souhaité.
Phosphorylation : Les extrémités 5' des linkers sont phosphorylées (avec de l'ATP et de la polynucléotide kinase). Ceci est important car la ligase a besoin d'un groupe phosphate 5' pour catalyser la formation de la liaison phosphodiester.
Ligation : Les linkers sont ligaturés aux extrémités du fragment d'ADN à cloner (utilisant de la DNA ligase).
Digestion : Le fragment d'ADN portant les linkers est digéré avec l'enzyme de restriction correspondant au site présent dans le linker. Ceci crée des extrémités cohésives compatibles avec le vecteur.
adapteur
Choix des adapteurs
Hybridation ou ligature
Appariement : Les fragments d'ADN portant les adapteurs sont mis en contact. Si les adapteurs ont été conçus correctement, leurs séquences complémentaires s'apparieront, ce qui permettra de rapprocher les fragments d'ADN.
Ligation
Avantages :
Universalité : N'importe quel fragment d'ADN peut être cloné grâce à l'utilisation de sites de restriction.
Inconvénients :
Methylation : Si le fragment d'ADN contient déjà un site de restriction pour la même enzyme que celle du linker, il doit être méthylé au préalable pour éviter qu'il ne soit coupé lors de la digestion
[DESSIN linker et adapteurs]
2. introduction du plasmide dans la bactérie “transformation”
Transformation bactérienne :
Principe : Les bactéries sont rendues compétentes pour absorber l'ADN exogène.
Méthodes :
Choc thermique : Les bactéries sont incubées dans une solution de chlorure de calcium, puis soumises à un choc thermique brutal (passage rapide de 0°C à 42°C). Ce choc thermique crée des pores transitoires dans la membrane bactérienne, permettant à l'ADN de pénétrer dans la cellule.
Électroporation : Les bactéries sont soumises à un champ électrique intense et bref, ce qui crée des pores dans la membrane bactérienne et permet à l'ADN de pénétrer.
Transformation de levures :
Principe : Les levures sont rendues perméables à l'ADN exogène.
Méthodes :
Traitement au lithium acétate : Les levures sont incubées avec du lithium acétate, ce qui fragilise leur paroi cellulaire et facilite l'entrée de l'ADN.
Électroporation : Comme pour les bactéries, l'électroporation peut être utilisée pour transformer les levures.
Transfection de cellules animales :
Principe : L'ADN exogène est introduit dans le noyau des cellules animales.
Méthodes :
Transfection chimique : Des agents chimiques (comme le chlorure de calcium ou des lipides cationiques) sont utilisés pour former des complexes avec l'ADN, ce qui facilite son entrée dans les cellules.
Électroporation : L'électroporation peut également être utilisée pour transfecter les cellules animales.
Microinjection : L'ADN est directement injecté dans le noyau de la cellule à l'aide d'une microaiguille.
Transduction virale : Un virus modifié est utilisé pour introduire l'ADN dans les cellules.
Transformation de cellules végétales:
Principe : L'ADN exogène est introduit dans les cellules végétales, souvent par l'intermédiaire de la bactérie Agrobacterium tumefaciens.
Méthodes :
Agrobacterium tumefaciens : Cette bactérie est naturellement capable de transférer de l'ADN (appelé ADN-T) dans les cellules végétales. L'ADN recombinant est inséré dans l'ADN-T, puis la bactérie est utilisée pour infecter les cellules végétales.
Bombardement de particules : Des microparticules d'or ou de tungstène sont recouvertes d'ADN, puis projetées à haute vitesse sur les cellules végétales, ce qui permet à l'ADN de pénétrer dans les cellules.
Électroporation : L'électroporation peut également être utilisée pour transformer les cellules végétales.
Facteurs à considérer pour choisir une méthode de transformation
Type de cellule hôte
taille et nature de l’ADN exogène
efficacité de la transformation
coût et facilité d’utilisation
il faut ensuite séléctionner seulement les bactéries qui ont accepté l’ADN exogène, Le mélange est étalé sur un milieu nutritif permettant une sélection des colonies contenant le DNA recombinant
3. laisse les multiplications se faire
4. isolation des plasmides avec ADN recombiné qui ont été produit
lyse cellulaire avec détergent SDS (change le pH)
Neutralisation avec un tampon pour inactiver les enzymes, cela précipite l’ADN chromosomique mais l'ADN plasmidique, plus petit et circulaire, reste en solution
Clarification
La solution est centrifugée pour séparer le précipité contenant l'ADN chromosomique et les débris cellulaires de la solution contenant l'ADN plasmidique.
Le surnageant contenant l'ADN plasmidique est récupéré.
caractéristiques essentielles d'un vecteur de clonage idéal
Indépendance du génome de la cellule hôte, il doit pouvoir se répliquer indépendamment
Capacité à accepter l'ADN passager : Le vecteur doit posséder un ou plusieurs sites de restriction uniques, qui quand ils sont ouverts n’affectent pas la capacité du plasmide à se répliquer
Introduction efficace dans la cellule hôte
Présence d'un gène marqueur (comme résistance aux antibiotiques) pour pouvoir ensuite faire la séléction
Nombre de copies élevé dans la cellule hôte
Facilité d'isolement de l'ADN du vecteur à la fin du processus
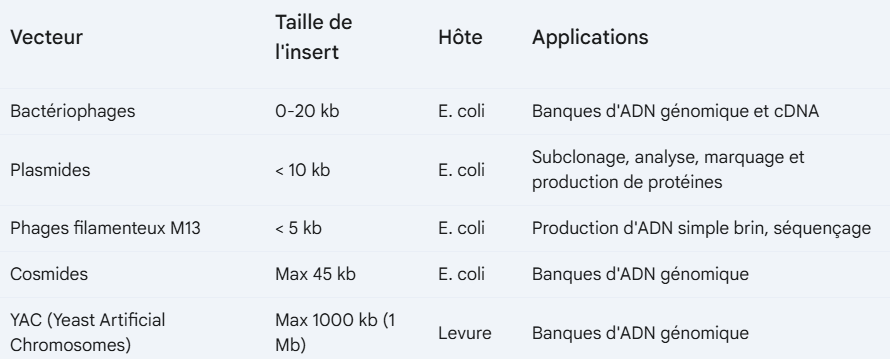
plasmides
Il existe plusieurs types de plasmides
Plasmides conjugatifs (facteurs F) : Portent les gènes nécessaires à la conjugaison, permettant le transfert de matériel génétique entre bactéries.
la conjugaison se fait d’une bactérie donneuse (qui a un plasmide F et un pilis sexuel) jusqu’à une bactérie récéptrice qui n’a aucun des deux. Il y a un transfert horizontal de plasmides et maintenant la deuxième bactéries possède un pili et un nouveau plasmide. Ce plasmide contient souvent aussi des gènes de résistance aux antibiotiques.
Plasmides de résistance (facteurs R) : Confèrent une résistance à un ou plusieurs antibiotiques ou à d'autres agents antimicrobiens.
le plasmide pBR322
un vecteur de clonage couramment utilisé en biologie moléculaire. Il a plusieurs caractéristiques importantes:
Origine de réplication (ori)
Gènes de résistance aux antibiotiques:
ampR
tetR
Sites de restriction uniques
le plasmide pUC19
c’est un dérivé de pBR322 mais il est plus petit. Il a des caractéristiques importantes:
Origine de réplication (ori)
Gènes de résistance aux antibiotiques:
ampR
Gène lacZ: Code pour la β-galactosidase,une enzyme qui clive le X-Gal en un produit bleu. C’est le gène marqueur qui montre qui a intégré le vecteur
colonie blanc/bleu → dans le gène lacZ il y a un site d’insertion et si l’insertion fonctionne, l’enzyme n’est plus fonctionnelle, ça aide à savoit si on a réussi à introduire le gène dans le plasmide
Colonies bleues = plasmide sans insert
Colonies blanches = plasmide avec insert
Site de clonage multiple (MCS)
Qu'est-ce qu'un vecteur d'expression ?
Un vecteur d'expression est un type de plasmide conçu pour permettre l'expression efficace d'un gène étranger dans une cellule hôte, généralement une bactérie comme E. coli. Il contient les éléments nécessaires pour la transcription et la traduction du gène inséré, ce qui permet la production de la protéine correspondante
Éléments clés du vecteur d'expression
Promoteur T7
voir promoteur inductible
Site de liaison du ribosome (boîte de Shine-Dalgarno)
Séquence codante
Site de clonage multiple (MCS) ou Polylinker
Étiquette 6-His et étiquette myc
peuvent être fusionnées à la protéine recombinante. Elles facilitent la purification et la détection de la protéine.
voir les tag après
Terminateur de transcription
Origine de réplication
Gène de résistance à l'ampicilline
confère une résistance permettant de sélectionner les bactéries ayant incorporé le plasmide.
Represseur lacI et site de liaison lacO
Le gène lacI code pour le répresseur LacI, qui se lie au site lacO (opérateur lactose) en l'absence d'inducteur (comme l'IPTG). La liaison du répresseur LacI bloque la transcription. En présence d'IPTG, le répresseur se détache du site lacO, permettant la transcription du gène sous le contrôle du promoteur T7.
Comment fonctionne un vecteur d'expression ?
Clonage : Le gène étranger est inséré dans le MCS du vecteur d'expression.
Transformation : Le vecteur recombinant est introduit dans des bactéries E. coli qui expriment l'ARN polymérase T7.
Induction : L'expression du gène étranger est induite par l'ajout d'IPTG, qui inactive le répresseur LacI et permet la transcription par l'ARN polymérase T7.
Traduction : Les ribosomes se lient à l'ARNm et traduisent la séquence codante en protéine.
Purification (facultatif) : La protéine recombinante peut être purifiée à l'aide des étiquettes (6-His ou myc).
promoteur inductible
De nombreuses protéines sont toxiques pour la cellule hôte, en particulier lorsqu'elles sont exprimées à des niveaux élevés. Pour éviter cet inconvénient, on utilise un promoteur inductible qui permet de contrôler l'expression du gène et de ne l'activer qu'au moment voulu.
ce qu’il contient:
Promoteur : Séquence d'ADN où se fixe l'ARN polymérase pour initier la transcription.
Séquence lacO : Séquence d'ADN où se fixe le répresseur LacI.
Gène d'intérêt : Séquence d'ADN codant pour la protéine que l'on souhaite exprimer.
Gène du répresseur LacI : Gène codant pour la protéine répresseur LacI.
Molécule inductrice IPTG: Analogue du lactose qui inactive le répresseur LacI.
comment ça marche:
En l'absence d'IPTG : Le répresseur LacI est produit et se fixe à la séquence lacO, empêchant l'ARN polymérase de transcrire le gène d'intérêt. L'expression du gène est bloquée.
En présence d'IPTG : L'IPTG se lie au répresseur LacI, ce qui l'empêche de se fixer à la séquence lacO. L'ARN polymérase peut alors transcrire le gène d'intérêt, permettant l'expression de la protéine correspondante.
TAGS - étiquettes
courtes séquences peptidiques ou protéiques qui sont fusionnées à la protéine recombinante lors du clonage. Ces étiquettes permettent de faciliter la purification, la détection ou la manipulation de la protéine recombinante.
Purification : Les étiquettes peuvent se lier à des matrices d'affinité spécifiques, ce qui permet de purifier la protéine recombinante à partir d'un mélange complexe.
Détection : Les étiquettes peuvent être reconnues par des anticorps spécifiques, ce qui facilite la détection de la protéine recombinante par des techniques comme le Western blot ou l'immunofluorescence.
Manipulation : Les étiquettes peuvent être utilisées pour immobiliser la protéine recombinante sur un support solide, ce qui facilite les études d'interaction ou d'activité enzymatique.
les étiquettes souvent utilisées sont myc (reconnue par des anticorps anti-myc spécifiques) et 6 his (se lie fortement aux ions nickel). Afin de séparer la protéine produite de la matrice avec laquelle on l’attrappe on fait une élution: A solution containing a high concentration of a substance that competes with the affinity tag for binding to the ligand is passed through the column. This "elutes" (releases) the recombinant protein from the ligand, and it is collected
baculovirus
Les baculovirus sont des virus qui infectent les insectes. Ils sont utilisés comme vecteurs d'expression car :
Ils peuvent infecter efficacement les cellules d'insectes.
Ils permettent l'expression de grandes quantités de protéines recombinantes.
Ils sont relativement sûrs car ils ne peuvent pas infecter les cellules de mammifères
Comment ça marche ?
Clonage du cDNA : Le cDNA codant pour la protéine d'intérêt est cloné dans un plasmide flanqué de séquences de baculovirus.
Cotransfection : Le plasmide contenant le cDNA et l'ADN de baculovirus linéarisé sont cotransfectés dans des cellules d'insectes.
Recombinaison : Par recombinaison homologue, le cDNA est inséré dans le génome du baculovirus, créant un baculovirus recombinant.
Infection : Les cellules d'insectes sont infectées avec le baculovirus recombinant.
Expression : Les cellules infectées expriment la protéine recombinante en grande quantité.
Avantages de l'utilisation des baculovirus :
Expression élevée
Modifications post-traductionnelles : Les cellules d'insectes sont capables d'effectuer les modifications post-traductionnelles nécessaires au bon fonctionnement des protéines eucaryotes (glycosylation, phosphorylation, etc.).
Correctement repliées : Les protéines recombinantes sont généralement correctement repliées
Inconvénients de l'utilisation des baculovirus :
Nécessite des cellules d'insectes
Peut prendre du temps
pCL-neo
pCI-neo : un vecteur d'expression pour les cellules de mammifères, plasmide conçu pour être maintenu comme un épisome (ADN extrachromosomique) dans les cellules de mammifères exprimant l'antigène grand T du virus SV40
contient plusieurs éléments importants
Enhancer/Promoteur CMV I.E.
Intron chimérique β-globine/IgG : Cet intron situé en aval du promoteur peut augmenter davantage l'expression du gène.
Signal de polyadénylation tardif SV40 : Ce signal permet d'augmenter les niveaux d'ARN stable.
Gène de la néomycine phosph transférase : Ce gène confère une résistance à la néomycine (G418), un marqueur de sélection pour les cellules de mammifères.
Origine de réplication (ori)
Origine de réplication f1
Promoteur T7 RNA polymérase : Permet la synthèse d'ARN in vitro.
Sites de restriction uniques
Comment fonctionne pCI-neo ?
Clonage : Le gène d'intérêt est inséré dans le MCS du plasmide pCI-neo.
Transfection : Le plasmide recombinant est transfecté dans des cellules de mammifères exprimant l'antigène grand T du SV40.
Expression : Le promoteur CMV I.E. permet l'expression constitutive du gène d'intérêt.
Sélection : Les cellules ayant intégré le plasmide sont sélectionnées en utilisant du G418.
Avantages de pCI-neo :
Expression constitutive : Permet une expression constante du gène d'intérêt.
Niveaux d'expression élevés : L'association du promoteur CMV I.E., de l'intron et du signal de polyadénylation permet d'obtenir des niveaux d'expression élevés.
Maintien épisomique : Le plasmide est maintenu comme un épisome dans les cellules exprimant l'antigène grand T, ce qui augmente l'expression.
Marqueur de sélection : Le gène de résistance à la néomycine permet de sélectionner les cellules transfectées.
Inconvénients de pCI-neo :
Nécessite l'expression de l'antigène grand T : Le plasmide est plus efficace dans les cellules exprimant l'antigène grand T du SV40.
résumé
Vecteur d'expression permet l'expression du gène codant pour la protéine d'intérêt. Il contient:
Un promoteur inductible : permet de contrôler l'expression du gène et de ne la déclencher qu'au moment voulu.
Des séquences régulatrices : contrôlent l'initiation et la terminaison de la transcription et de la traduction.
Un gène de résistance à un antibiotique : permet de sélectionner les cellules ayant intégré le plasmide.
on peut infecter différentes sorte de cellules avec (bactérie, levure, insecte, mammifère)
Il faut mettre des étiquettes sur les protéines produites
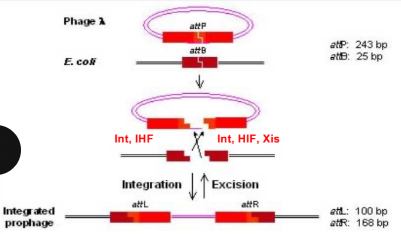
phage lamda
Virus qui infecte les bactéries de la famille des Siphoviridae. Peut soit faire un cylce lytique soit lysogénique
l’avantage c’est qu’on peut introduire des gènes plus grands et que l’infection est très efficace
Plaque Assay: comment compter les phages
dans cycle lysogénique
recombinaison spécifique → intégration et l'excision du phage dans le chromosome bactérien.
Sites d'attachement : attP et attB
attP : Site d'attachement sur l'ADN du phage lambda.
attB : Site d'attachement sur le chromosome bactérien de E. coli.
Intégration et excision : deux réactions réversibles
Intégration :
Réaction entre attP et attB.
Formation de deux nouveaux sites : attL et attR.
Nécessite les protéines Intégrase (Int) et Facteur d'intégration de l'hôte (IHF).
Excision :
Réaction entre attL et attR.
Restauration des sites attP et attB.
Nécessite les protéines Intégrase (Int), Facteur d'intégration de l'hôte (IHF) et Excisionase (Xis).
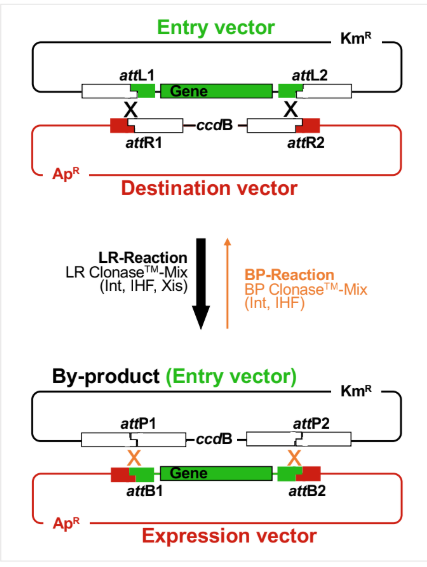
insertion du gène voulu dans la bactérie
Système gateway
Les différents vecteurs :
Vecteur d'entrée (Entry vector) : Contient l'insert flanqué de sites attL1 et attL2. Il est résistant à la kanamycine (KmR).
Vecteur de destination (Destination vector) : Contient les sites attR1 et attR2, ainsi qu'un gène de sélection négative ccdB. Il est résistant à l'ampicilline (ApR).
Vecteur d'expression (Expression vector) : Le vecteur final contenant l'insert et les éléments nécessaires à son expression. Il est obtenu après recombinaison entre le vecteur d'entrée et le vecteur de destination.
→ c’est pour mettre le gène qui nous intéresse dans le phage
avantages:
Efficacité et rapidité
Polyvalence
Le système Gateway permet de cloner un même insert dans différents vecteurs (d'expression, de destination, etc.) sans avoir à le subcloner à chaque fois. Il suffit de réaliser une seule réaction de recombinaison pour transférer l'insert d'un vecteur à un autre.
Standardisation
Compatibilité
Réversibilité
clonage par recombinaison Gateway multisite
technique pour combiner plusieurs fragments d'ADN en une seule construction
objectif: créer un clone d'expression contenant plusieurs fragments d'ADN (Frag1, Frag2, Frag3) dans un ordre spécifique
étapes:
trouver la séquence du gène lsl-1
designing the oligos to amplify (by PCR) the different genomic regions for cloning into the entry vectors. Il y a la séquence sens et antisens. Il y a trois cone d’entrée
Il faut aussi mettre:
les régions att
le codon start
une partie de la séquence du gène lsl-1
codon stop
sites de restriction
une partie de la séquence 3’ UTR
il faut mettre dune étiquette au milieu de la séquence
réaction LR
Matériel de départ :
Vous avez trois clones d'entrée (5' Entry Clone, Middle Entry Clone, 3' Entry Clone), contenant chacun un fragment d'ADN spécifique (Isl-1 p, Isl-1 cds-FLAG, Isl-1 3'UTR) flanqué de sites attL.
Vous avez également un vecteur de destination appelé pCG150, contenant des sites attR.
Les sites attL sur les clones d'entrée réagissent avec les sites attR sur le vecteur de destination. Cela conduit à la recombinaison des fragments d'ADN dans le vecteur de destination, ce qui donne la construction finale.
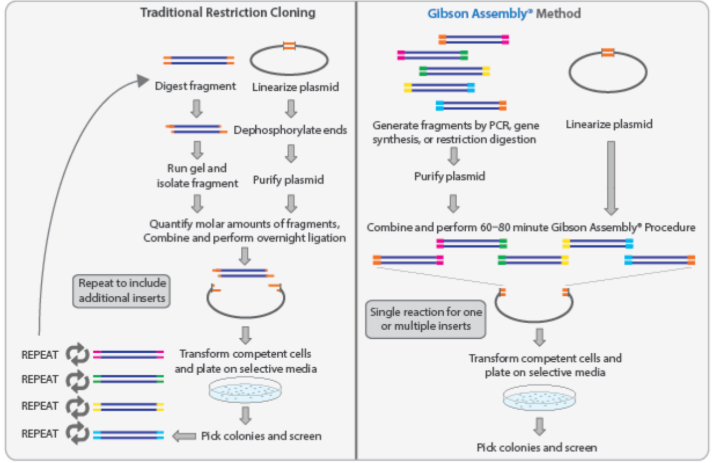
assemblage de Gibson
méthode de clonage génétique qui permet d'assembler plusieurs fragments d'ADN en un seul morceau d'ADN.
L'assemblage de Gibson repose sur l'utilisation d'une enzyme, la Gibson Assembly Master Mix, qui contient 3 activités enzymatiques :
Exonucléase: Elle dégrade l'ADN double brin à partir de l'extrémité 5', créant ainsi des extrémités simple brin cohésives.
ADN polymérase: Elle synthétise l'ADN complémentaire des extrémités simple brin cohésives, permettant ainsi l'appariement des fragments d'ADN.
ADN ligase: Elle ligue les fragments d'ADN entre eux, formant ainsi un seul morceau d'ADN.
Étapes de l'assemblage de Gibson
Conception des fragments d'ADN : Les fragments d'ADN à assembler doivent être conçus de manière à ce que leurs extrémités se chevauchent sur une vingtaine de bases. Ces chevauchements permettent aux fragments de s'apparier entre eux lors de la réaction d'assemblage.
Préparation des fragments d'ADN : Les fragments d'ADN peuvent être obtenus par PCR, par digestion enzymatique ou par synthèse chimique.
Réaction d'assemblage : Les fragments d'ADN sont mélangés avec la Gibson Assembly Master Mix et incubés à une température optimale pendant une durée déterminée.
Transformation bactérienne : Le produit de la réaction d'assemblage est transformé dans des bactéries compétentes.
Sélection des clones : Les bactéries contenant le plasmide recombinant sont sélectionnées sur un milieu de culture approprié.
Avantages de l'assemblage de Gibson
Simplicité
Efficacité : L'assemblage de Gibson est très efficace et permet d'obtenir un grand nombre de clones recombinants.
Flexibilité : La technique permet d'assembler un nombre quelconque de fragments d'ADN, ce qui la rend très flexible pour la construction de vecteurs complexes.
Rapidité : L'assemblage de Gibson est une technique rapide qui peut être réalisée en quelques heures.
Inconvénients de l'assemblage de Gibson
Coût : La Gibson Assembly Master Mix peut être coûteuse.
Conception des fragments d'ADN : La conception des fragments d'ADN peut être complexe, en particulier pour l'assemblage de nombreux fragments.
Applications de l'assemblage de Gibson
Assemblage de gènes
Construction de vecteurs d'expression
Création de banques d'ADN
Mutagenèse dirigée
comparaison clonage traditionel et assemblage de Gibson
Préparation de l’ADN génomique
on ne peut pas mettre n’importe quoi dans le phage,
on prend de l’ADN on le digère avec des enzymes de restrictions — on a plusieurs échantillons, avec plusieurs temps différent en contact de l’enzyme.
on fait ensuite une élécrophorèse pour les trier par taille et on prend que la bonne taille.
on fait un bras vecteur qui content plusieurs bout d’ADN ok
on fait des coupures concatemer pour mettre dans plusieurs phages différents
les phages infectent les bactéries
clonage et d'excision en utilisant le système Zap
on a un pBluescript SK-phagemid, vecteur qui peut exister à la fois sous forme de plasmide (ADN circulaire) et de phage (un virus qui infecte les bactéries). On met l’ADN cible dedans. on infecte des bactéries avec ce vecteur, multiplication et finalement libération des phages chacun portant une copie de votre plasmide pBluescript avec votre insert d'ADN.
interlocked circular DNA molecules
The problem:
Bacteriophage P1 DNA replicates in a way that can sometimes lead to the formation of interlocked circular DNA molecules.
These interlocked molecules (dimers) need to be resolved into individual circular DNA molecules for proper packaging and propagation of the phage.
The solution: Cre-loxP system
Cre recombinase: An enzyme that can cut and rejoin DNA at specific sequences called loxP sites.
loxP sites: Short DNA sequences that are recognized by the Cre recombinase
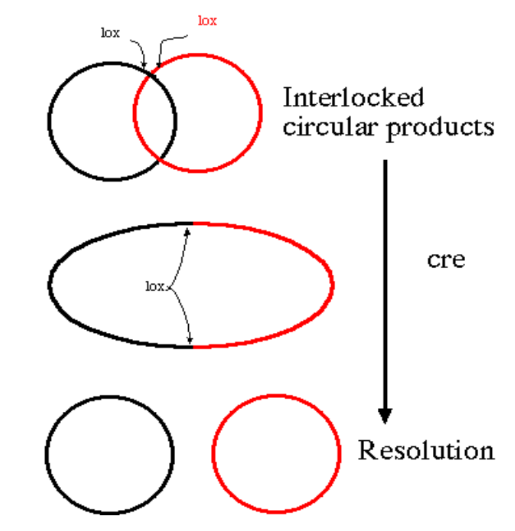
on peut utiliser ce système dans la modification génétique
Vous avez deux types de souris :
Une souris "Cre" qui possède une enzyme spéciale (Cre recombinase) capable de couper l'ADN à des endroits précis.
Une souris "LoxP" dont la recette (gène cible) est entourée de marques spéciales (sites loxP).
Vous croisez les deux souris :
Vous obtenez une souris qui possède à la fois l'enzyme Cre et les marques loxP autour de sa recette.
Vous activez l'outil spécial :
Vous donnez à la souris Cre un produit qui active l'enzyme Cre.
L'enzyme Cre coupe l'ADN aux endroits marqués (sites loxP), ce qui supprime une partie de la recette (gène cible).
La recette est modifiée !

cosmides
type de vecteur, ils combinent les avantages des plasmides (faciles à manipuler) et du bactériophage λ (peuvent transporter de grands inserts d'ADN).
Comment fonctionnent les cosmides
ADN génomique : L'ADN de l'organisme que vous souhaitez étudier est partiellement coupé en fragments d'environ 35-45 kb à l'aide d'une enzyme comme Sau3A.
Vecteur cosmide : Un vecteur plasmidique est linéarisé (ouvert) et ses extrémités sont compatibles avec les fragments d'ADN génomique.
Ligation : Les fragments d'ADN génomique sont insérés (ligués) dans le vecteur cosmide, créant de longues chaînes d'ADN appelées concatémères.
Emballage in vitro : Ces concatémères sont emballés dans des particules de phage à l'aide d'une technique appelée emballage in vitro. Ce processus reconnaît les sites cos sur l'ADN du cosmide et emballe l'ADN entre eux.
Infection : Les particules de phage infectent les bactéries E. coli.
Sélection : Les bactéries sont cultivées sur un milieu contenant de la tétracycline (tet), un antibiotique. Seules les bactéries contenant le cosmide (qui porte un gène de résistance à tet) survivront et formeront des colonies
Avantages des cosmides :
Efficacité de transformation élevée : Comme les plasmides, les cosmides peuvent être facilement introduits dans les bactéries.
Grands inserts d'ADN : Comme le bactériophage λ, les cosmides peuvent transporter de grands fragments d'ADN (40-45 kb), permettant le clonage de gènes entiers ou même d'opérons.
Nombre de copies élevé : Les cosmides peuvent se répliquer en grand nombre dans les bactéries, ce qui donne un bon rendement d'ADN.
YAC, BAC et PAC
YACs: Yeast artificial chromosomes are vectors that can carry even larger DNA inserts
les levures acceptent facilement un chromosome en plus
les cosmides et les YAC ensembles permettent de former une physical map génétique, shows the actual physical locations of DNA segments along a chromosome
structure de vecteur YAC
ARS (Séquence de Réplication Autonome) : Origine de réplication, permet au YAC de se répliquer dans la levure.
CEN (Centromère) : Assure la ségrégation correcte du YAC lors de la division cellulaire.
TEL (Télomères) : Protège les extrémités du YAC de la dégradation.
Sites EcoRI et BamHI : Sites d'enzymes de restriction utilisés pour insérer l'ADN étranger.
Marqueurs de sélection : Gènes qui permettent la sélection des cellules de levure contenant le YAC.
YAC : Avantages et inconvénients
Avantage : Les YAC peuvent transporter de très grands inserts d'ADN (jusqu'à 1 million de paires de bases), ce qui les rend utiles pour le clonage et la cartographie de grands génomes.
Inconvénients :
Difficiles à manipuler : L'introduction de YAC dans les cellules de levure (électroporation) peut être difficile.
Instabilité : Les YAC sont sujets à des réarrangements et à des recombinaisons, ce qui peut altérer la séquence d'ADN d'origine et la rendre différente de la séquence du génome source.
BAC (Bacterial Artificial Chromosomes)
PAC (Chromosomes Artificiels Dérivés de P1)
Pourquoi les BAC et les PAC sont préférés
E. coli comme hôte : Les BAC et les PAC utilisent la bactérie E. coli comme cellules hôtes, qui sont plus faciles à manipuler que la levure.
Faible nombre de copies : Les BAC et les PAC existent en très faible nombre de copies dans les cellules bactériennes. Cela aide à prévenir la recombinaison et à maintenir la stabilité de l'ADN cloné.
Différences entre les BAC et les PAC
BAC : Dérivés du facteur F de E. coli, qui a une origine de réplication strictement contrôlée.
PAC : De conception similaire aux BAC, mais utilisent l'origine de réplication du bactériophage P1, ce qui permet un cycle de réplication par génération bactérienne.
How BAC cloning works:
DNA insert preparation: The DNA to be cloned is cut into large fragments using restriction enzymes.
Ligation: The DNA fragments are inserted into the BAC vector that has been linearized using restriction enzymes.
Transformation: The BAC construct is introduced into E. coli cells via electroporation.
Selection: Bacteria containing the BAC are selected based on their resistance to chloramphenicol and counterselection against the sacB gene.
How PAC cloning works:
DNA insert preparation: The DNA to be cloned is cut into large fragments using restriction enzymes.
Ligation: The DNA fragments are inserted into the PAC vector that has been linearized using restriction enzymes.
Transformation: The PAC construct is introduced into E. coli cells.
Selection: Bacteria containing the PAC are selected based on their resistance to kanamycin and counterselection against the sacB gene.
préparation du DNA génomique pour clonage dans YAC, BAC et PAC
Extraction de l'ADN génomique
Fragmentation de l'ADN génomique
Sélection de la taille des fragments (facultatif)
Modification des extrémités des fragments (si nécessaire) avec adapteur
Ligature dans les vecteurs
Transformation ou transfection
Les vecteurs contenant les fragments d'ADN génomique sont introduits dans des cellules hôtes appropriées (levure pour les YAC, bactéries pour les BAC et PAC) par transformation ou transfection.
Sélection des clones