10 Protein breakdown
1/80
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
81 Terms
What are the two major sources of amino acids for the body?
Dietary protein digestion and breakdown of cellular proteins.
Where does dietary protein digestion begin?
Stomach.
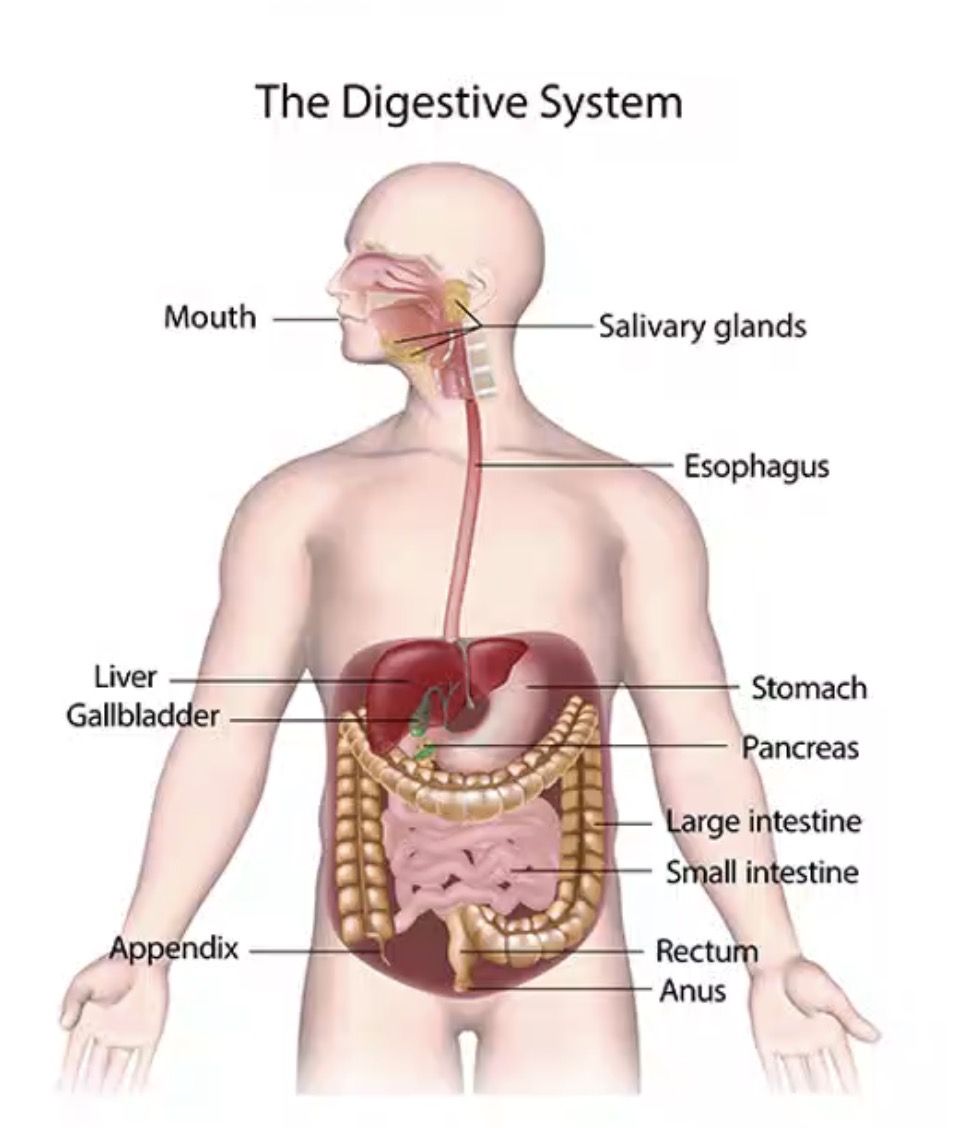
What enzyme begins protein digestion in the stomach?
Pepsin.
Why is pepsin effective in the stomach?
Acidic conditions denature proteins, making them easier to digest.
What happens to partially digested proteins after the stomach?
Further digestion in the small intestine.
What products are absorbed from protein digestion?
Single amino acids, dipeptides, and tripeptides.
Why are cellular proteins broken down?
To remove damaged/unwanted proteins and recycle amino acids.
What is protein turnover?
Continuous synthesis and degradation of proteins.
Why is protein turnover important?
Quality control, regulation, amino acid recycling, and adaptation.
How are unwanted intracellular proteins marked for destruction?
Ubiquitination.
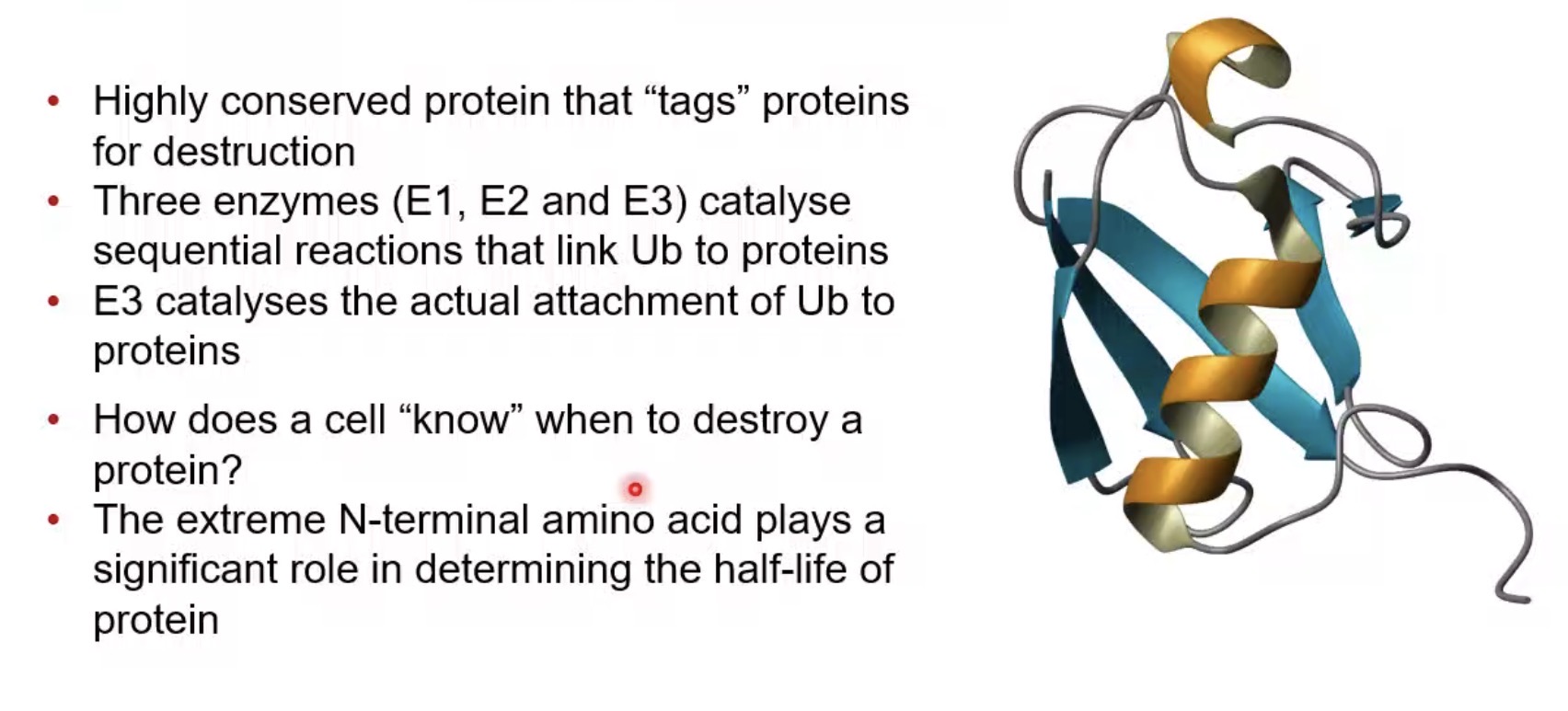
What is ubiquitin?
A small highly conserved protein that tags proteins for degradation.
What is the role of ubiquitin?
Marks proteins for destruction by the proteasome.
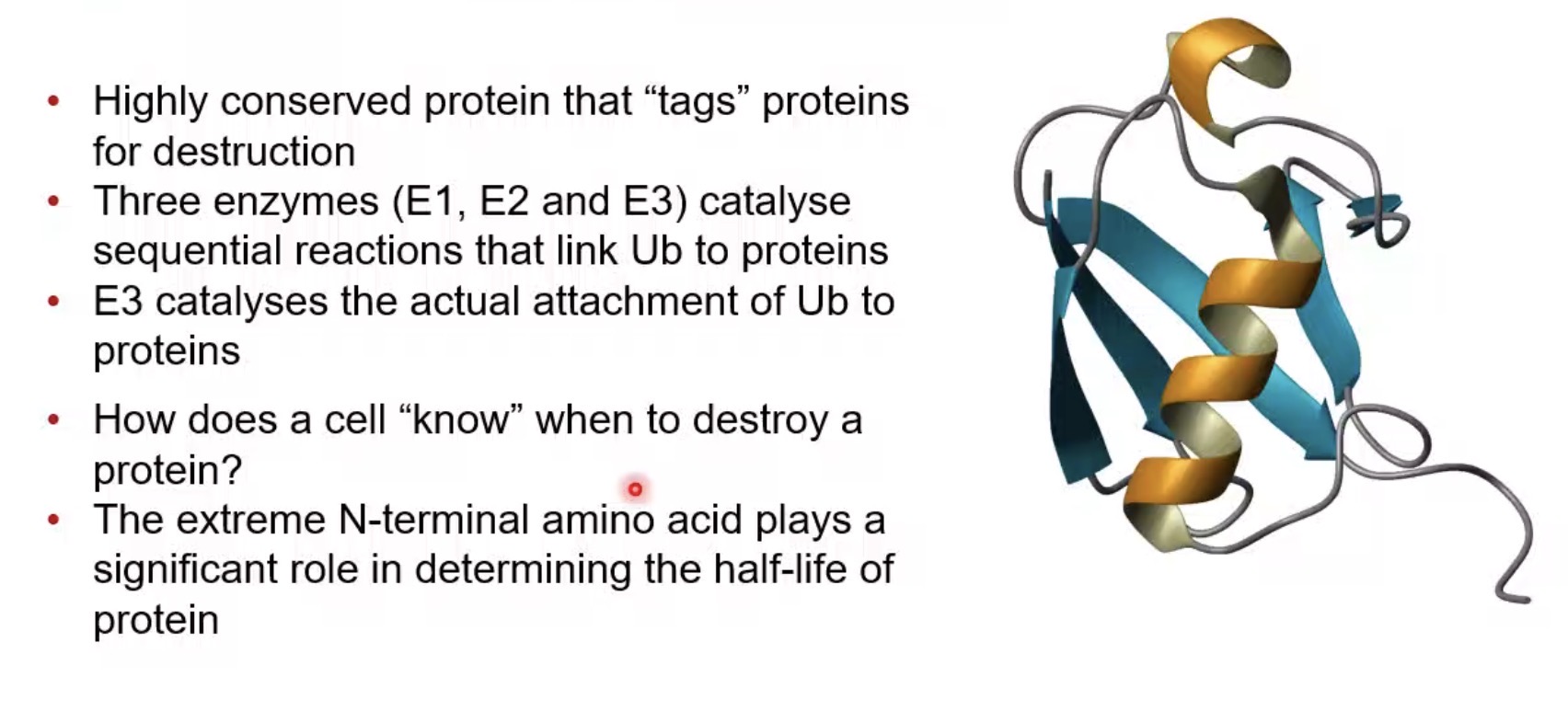
How many enzyme classes are involved in ubiquitination?
Three (E1, E2, E3).
What does E1 do in ubiquitination?
Activates ubiquitin.
What does E2 do in ubiquitination?
Carries activated ubiquitin.
What does E3 do in ubiquitination?
Transfers ubiquitin to the target protein.
Which ubiquitination enzyme determines substrate specificity?
E3 ligase.
Why is E3 important?
It decides which proteins are degraded.
What determines protein half-life in some proteins?
The N-terminal amino acid (N-end rule concept).
What is the proteasome?
A large protein complex that degrades ubiquitinated proteins.
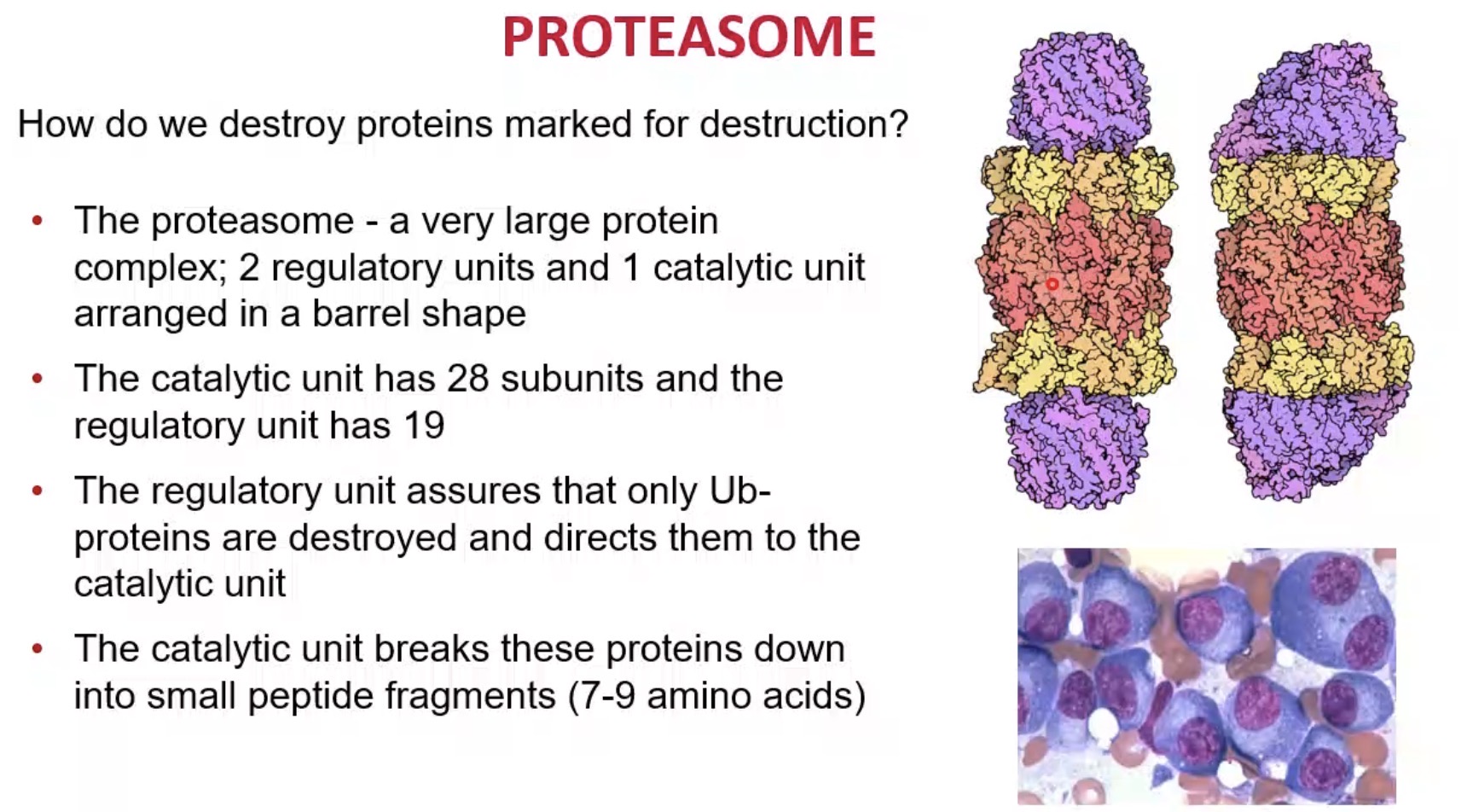
Structure of proteasome?
Barrel-shaped complex with regulatory and catalytic units.
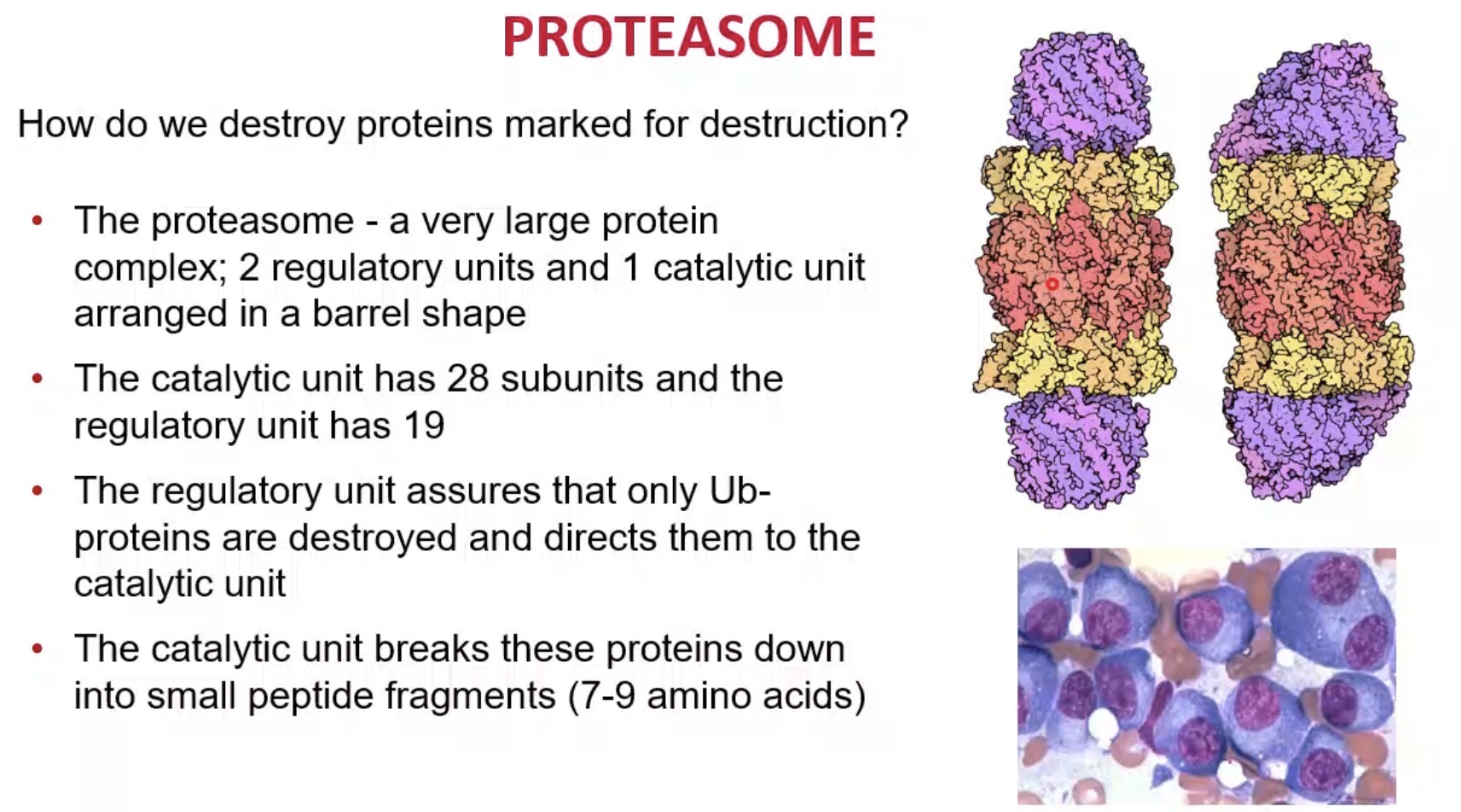
How many regulatory units does the proteasome have?
2.
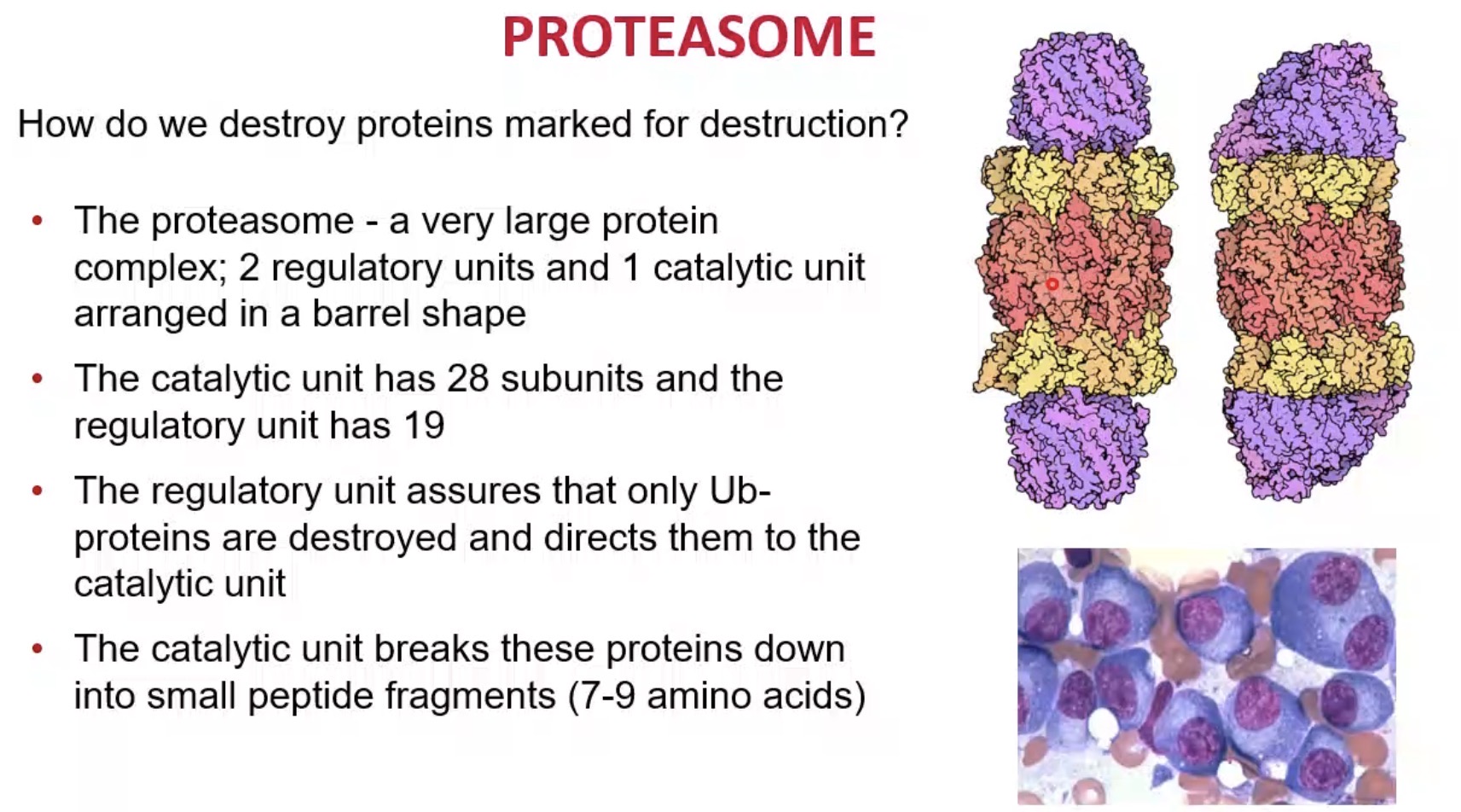
How many catalytic units does the proteasome core contain?
28 subunits.
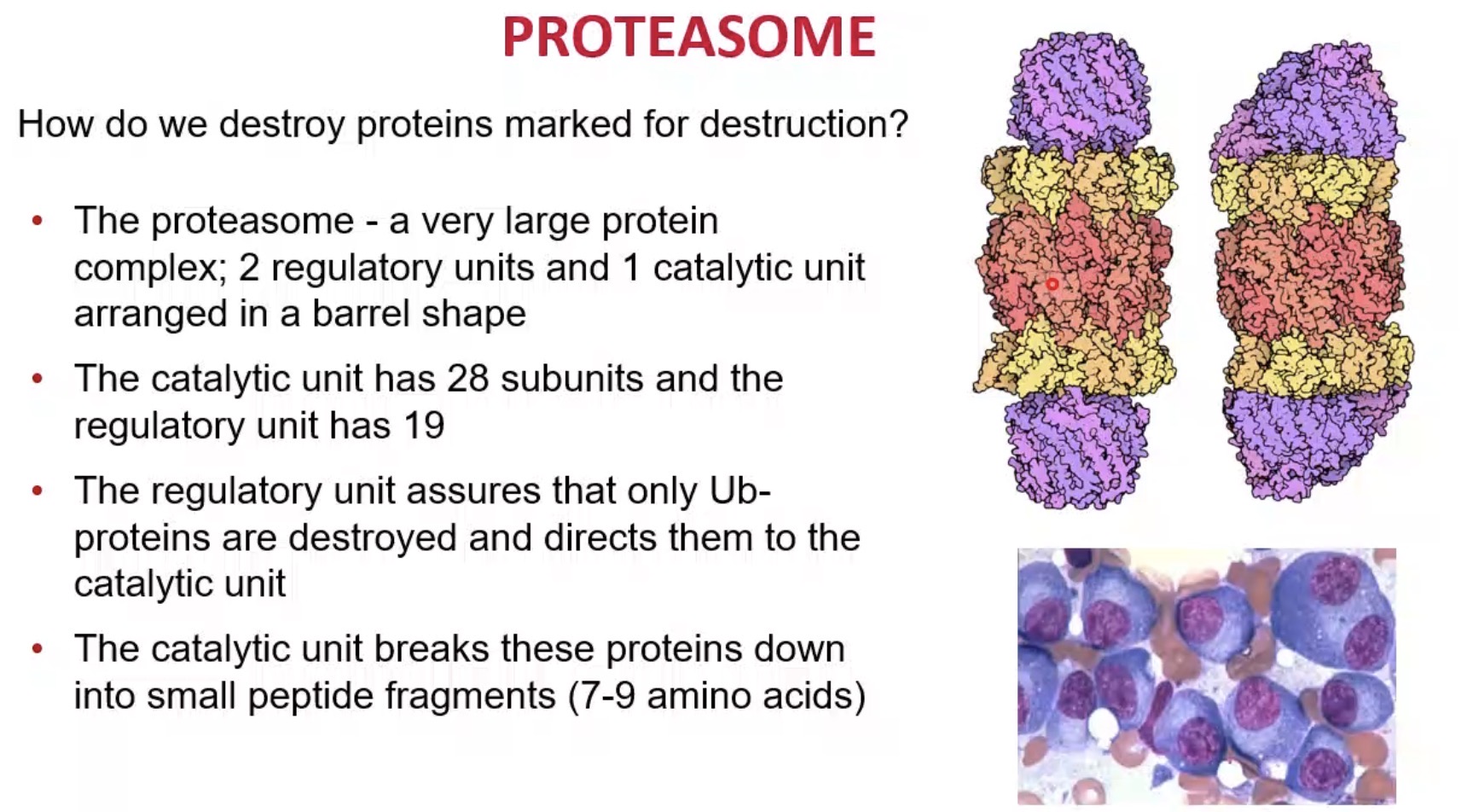
What is the role of proteasome regulatory units?
Recognise ubiquitinated proteins and direct them into the core.
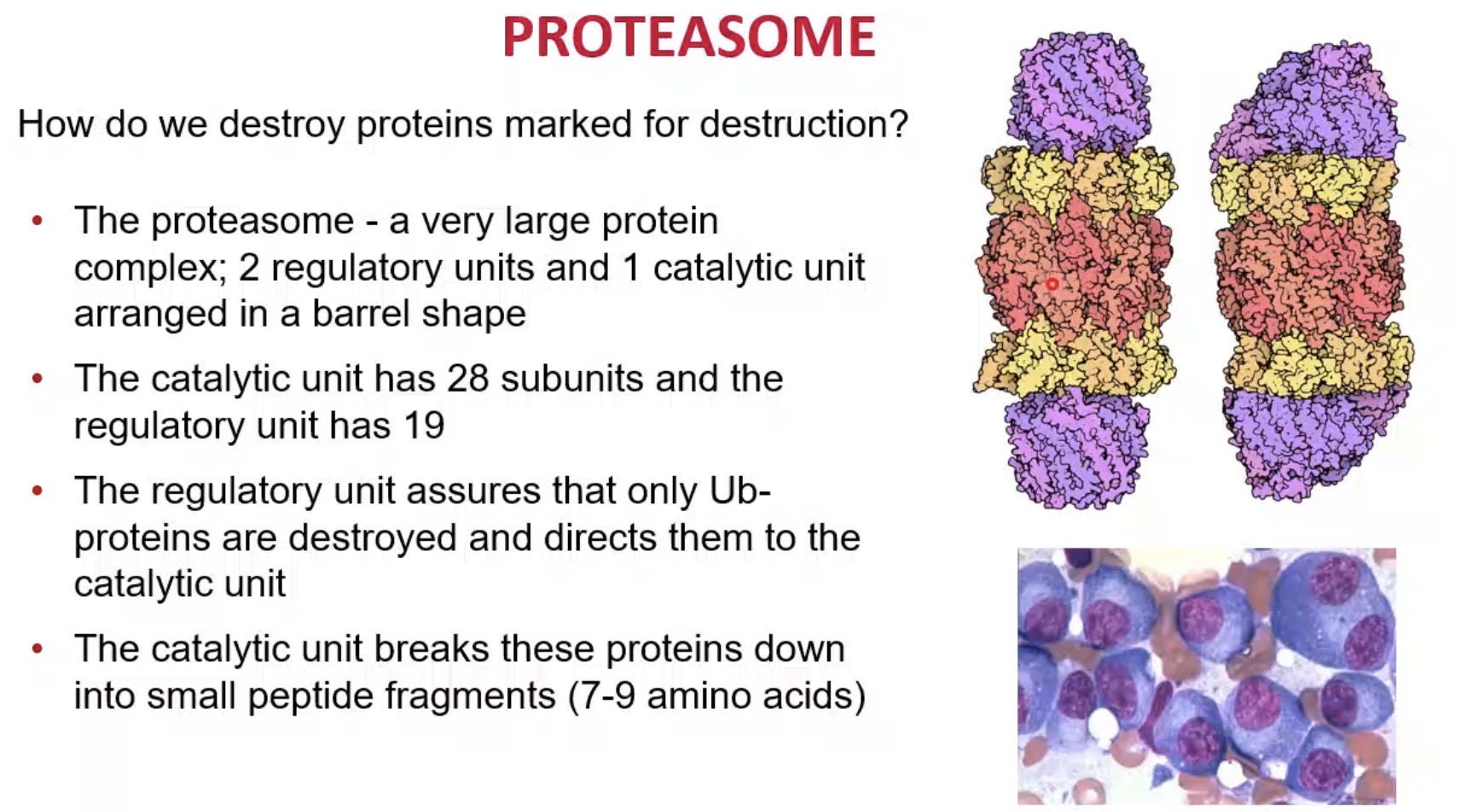
What is the role of the catalytic core?
Break proteins into peptide fragments.
What size peptides are typically produced by proteasomes?
Approximately 7–9 amino acids.
What happens to peptides after proteasomal degradation?
Further broken into amino acids.
What are the possible fates of amino acids?
Protein synthesis or metabolic breakdown.
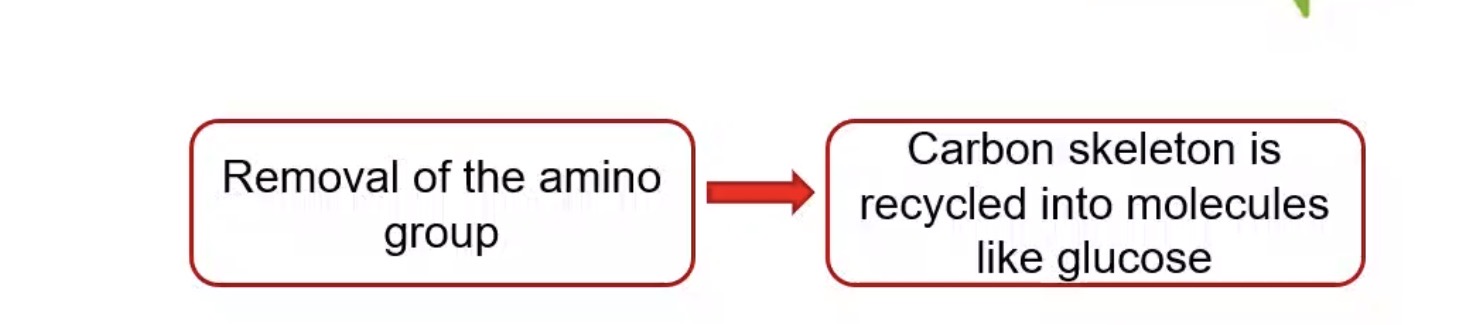
What is the first step in amino acid breakdown?
Removal of the amino group.
What enzymes commonly remove amino groups?
Aminotransferases.
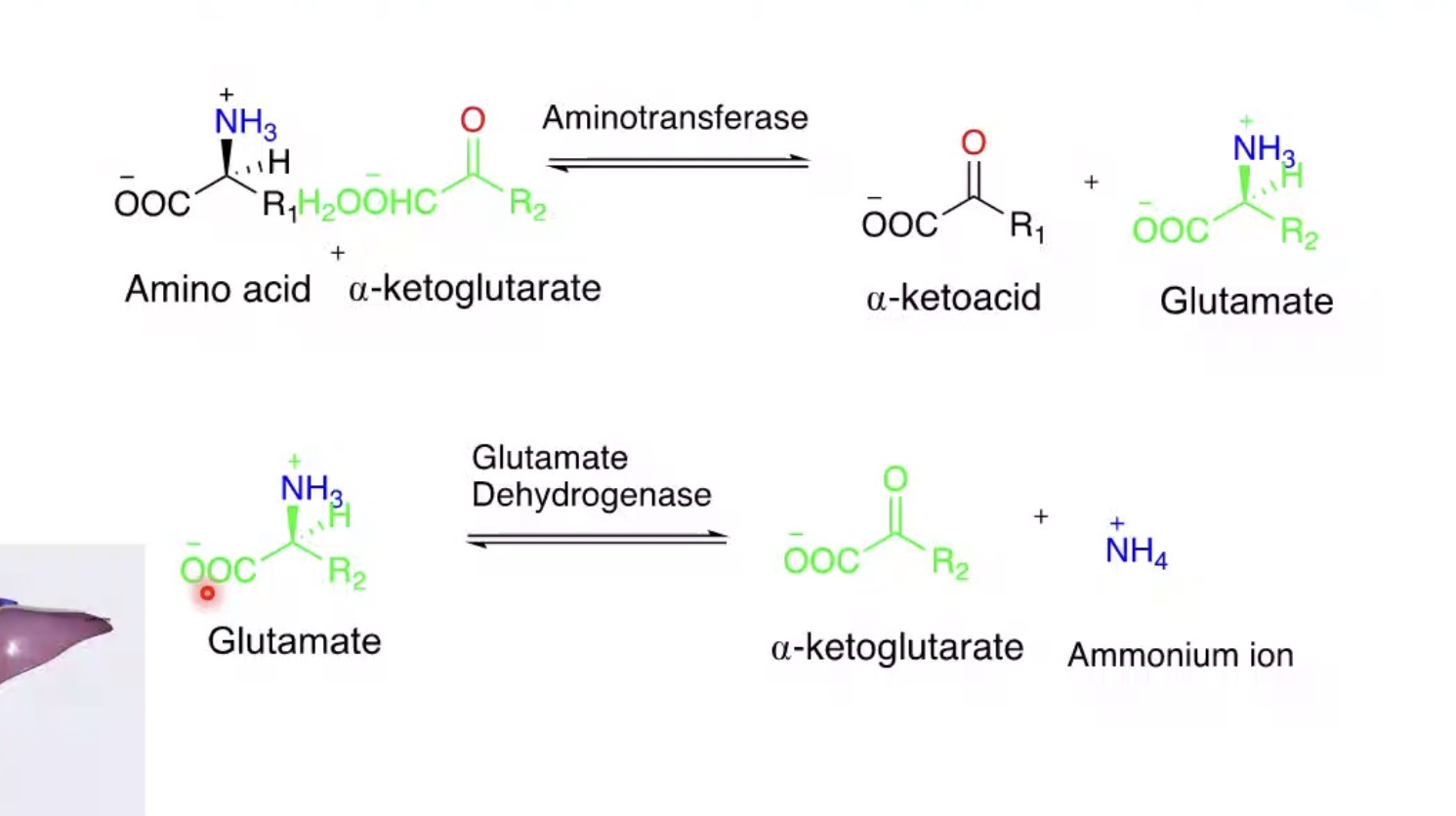
What is transamination?
Transfer of an amino group to another molecule.
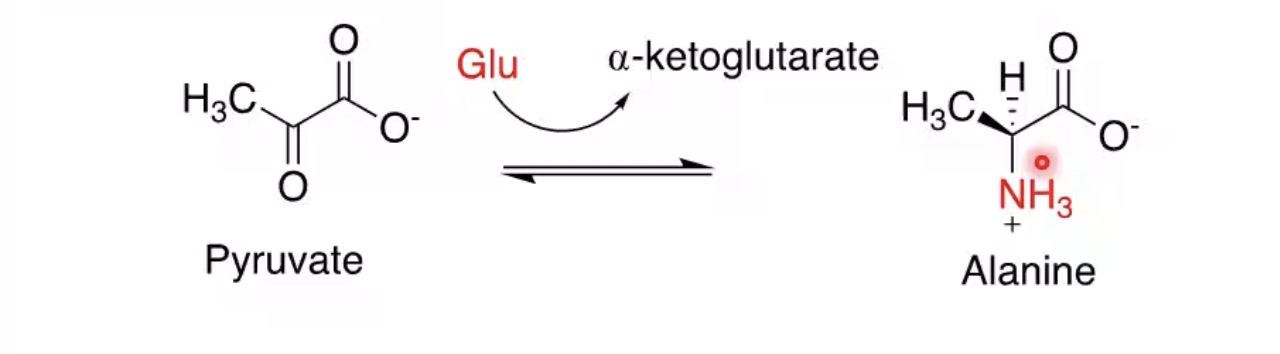
What molecule commonly receives amino groups?
α-ketoglutarate.
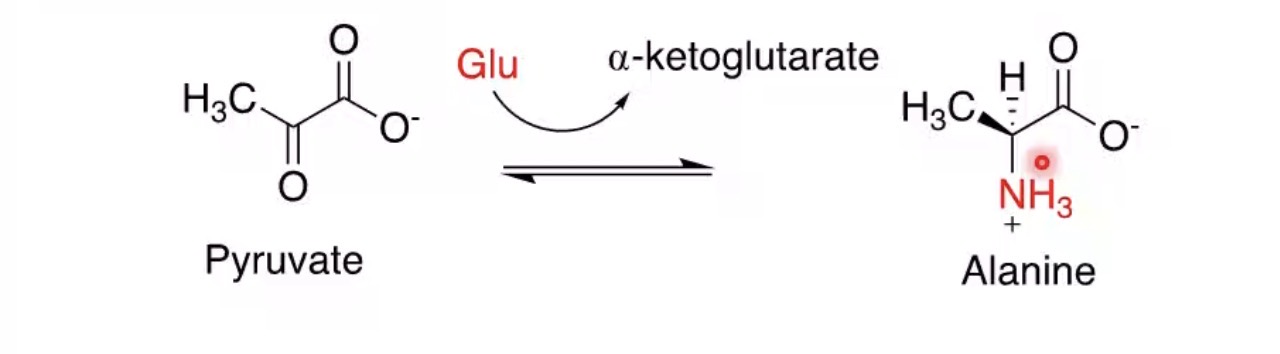
Product formed when α-ketoglutarate accepts an amino group?
Glutamate.
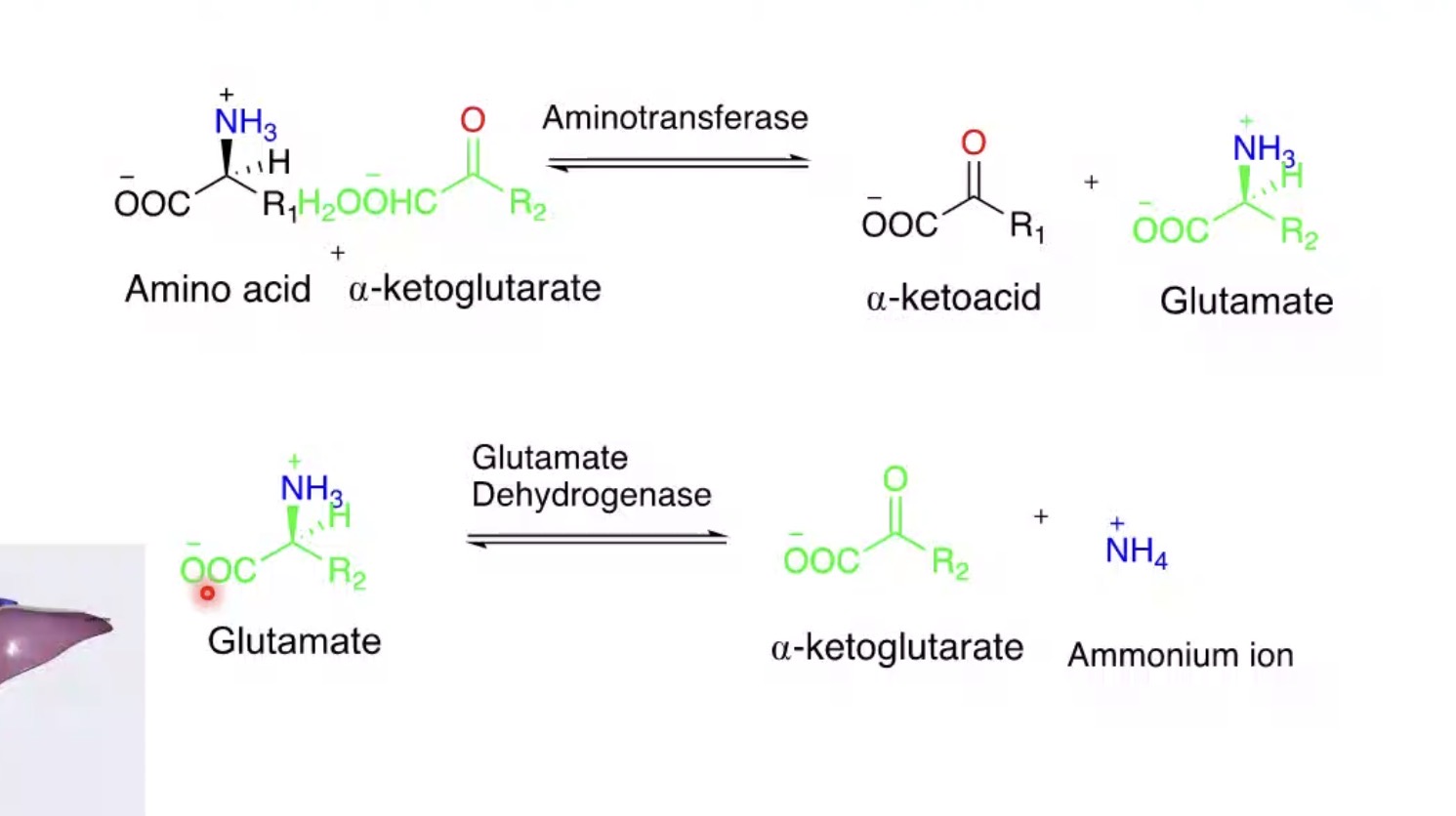
What happens to the amino acid after transamination?
Becomes an α-keto acid.
Why is glutamate central in amino acid metabolism?
Collects amino groups for disposal.
Can aminotransferases catalyse both directions?
Yes.
What is oxidative deamination?
Removal of amino group as ammonium.
Why is excess ammonium dangerous?
Toxic, especially to the nervous system.
What happens to excess ammonium?
Converted to urea.
Where does the urea cycle occur?
Liver.
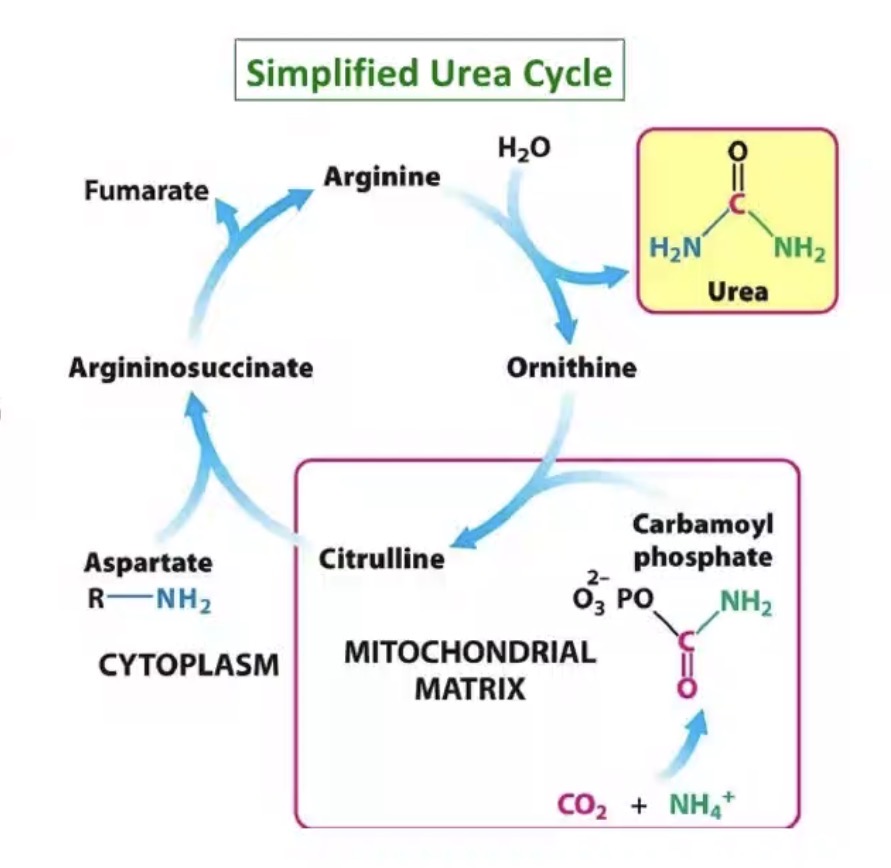
Why is the liver essential in nitrogen disposal?
Converts toxic nitrogen into excretable urea.
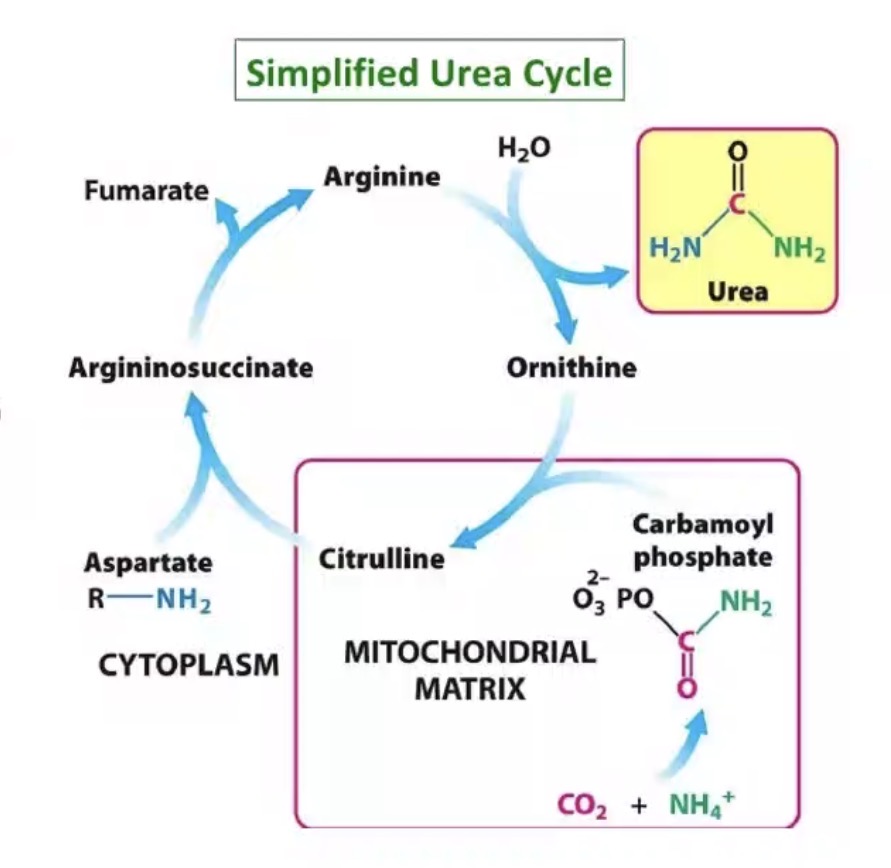
How many nitrogen atoms are in urea?
Two.
Sources of nitrogen in urea?
One from ammonia/ammonium, one from aspartate.
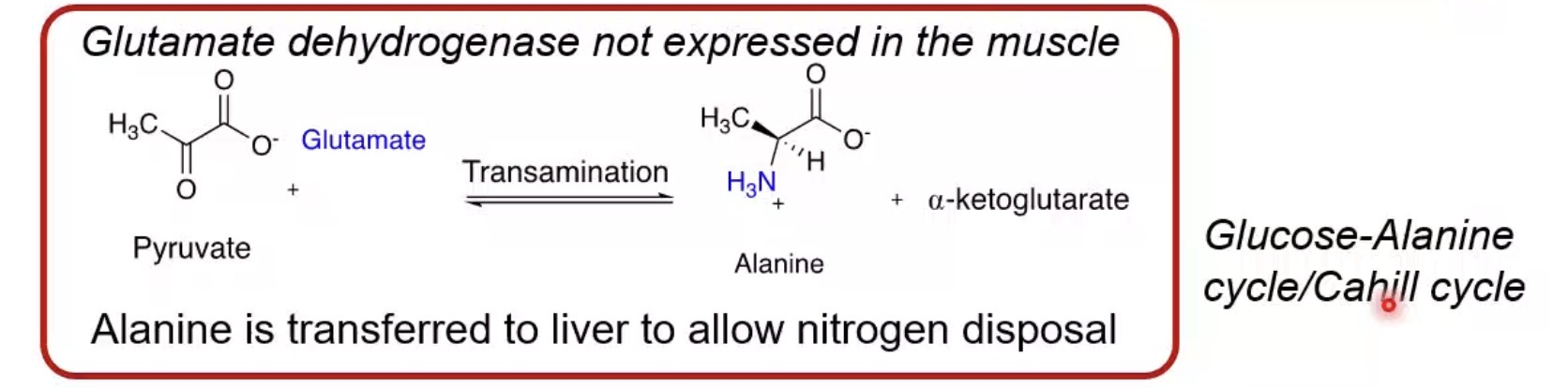
Can muscle perform the urea cycle?
No.
How does muscle handle excess nitrogen?
Transfers it as alanine to the liver.
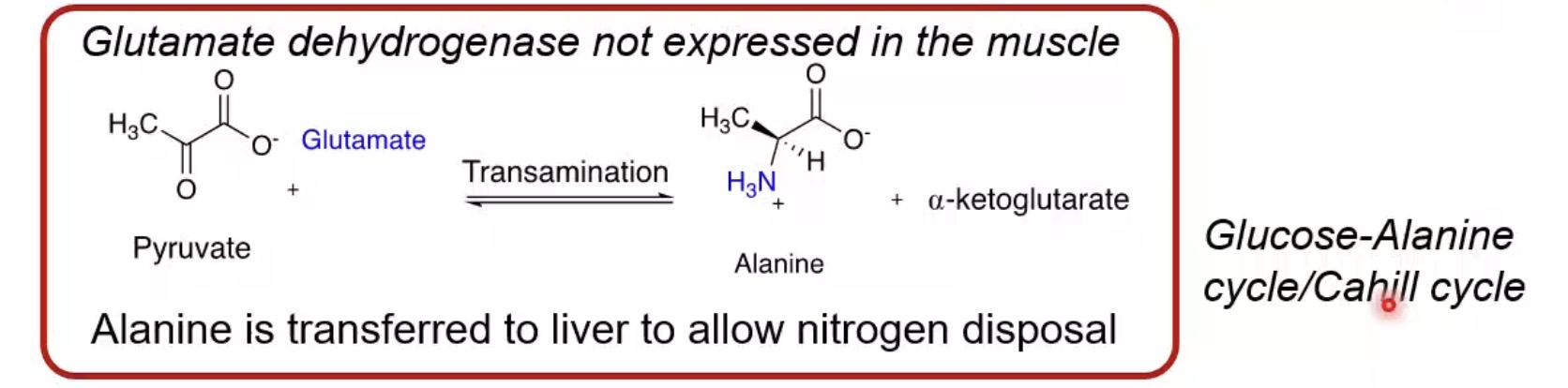
What is the glucose-alanine cycle?
Alanine transports amino nitrogen from muscle to liver.
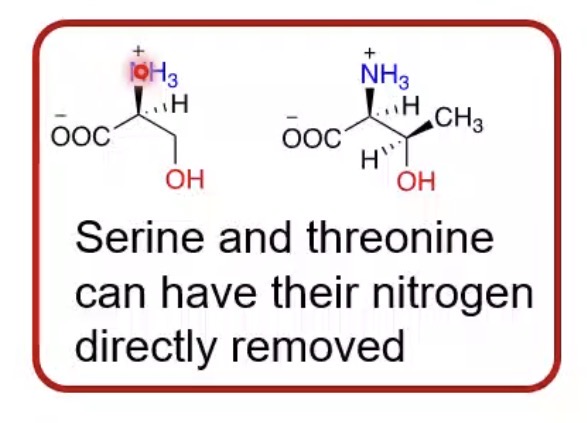
Which amino acids can lose nitrogen directly without transamination?
Serine and threonine.
What happens to amino acid carbon skeletons?
Recycled into metabolic intermediates.

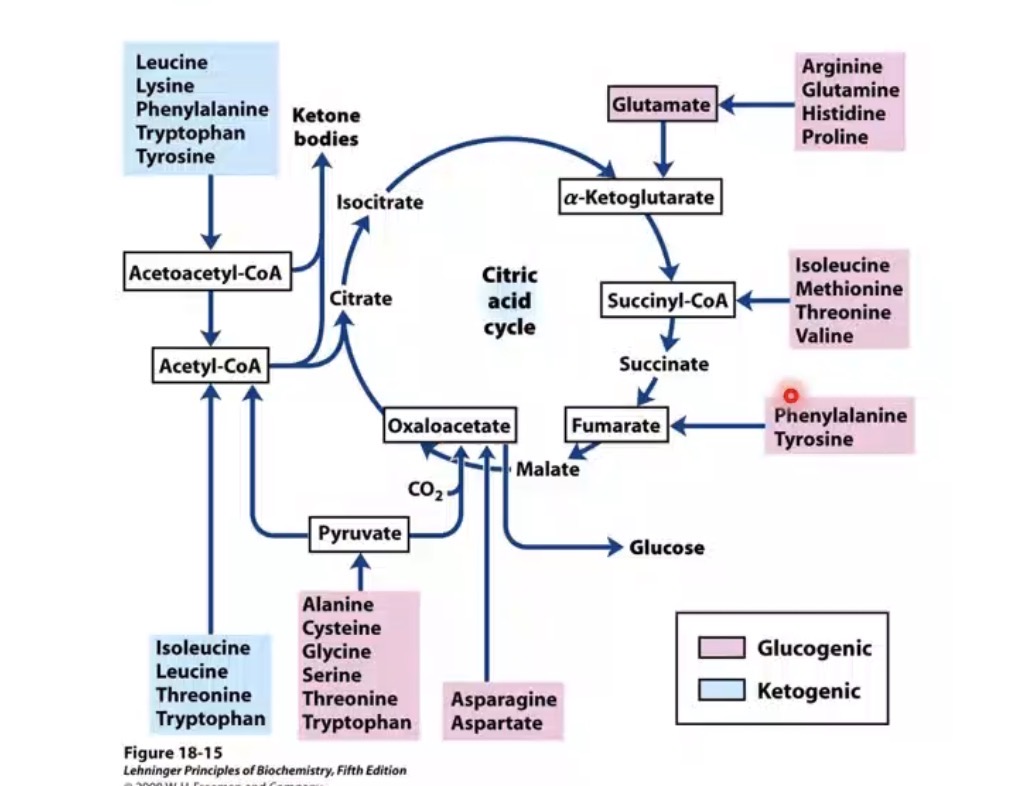
What are the two amino acid metabolic classes?
Glucogenic and ketogenic.
What are glucogenic amino acids?
Amino acids yielding glucose precursor intermediates.
What intermediates can glucogenic amino acids produce?
Pyruvate, α-ketoglutarate, succinyl CoA, fumarate, oxaloacetate.
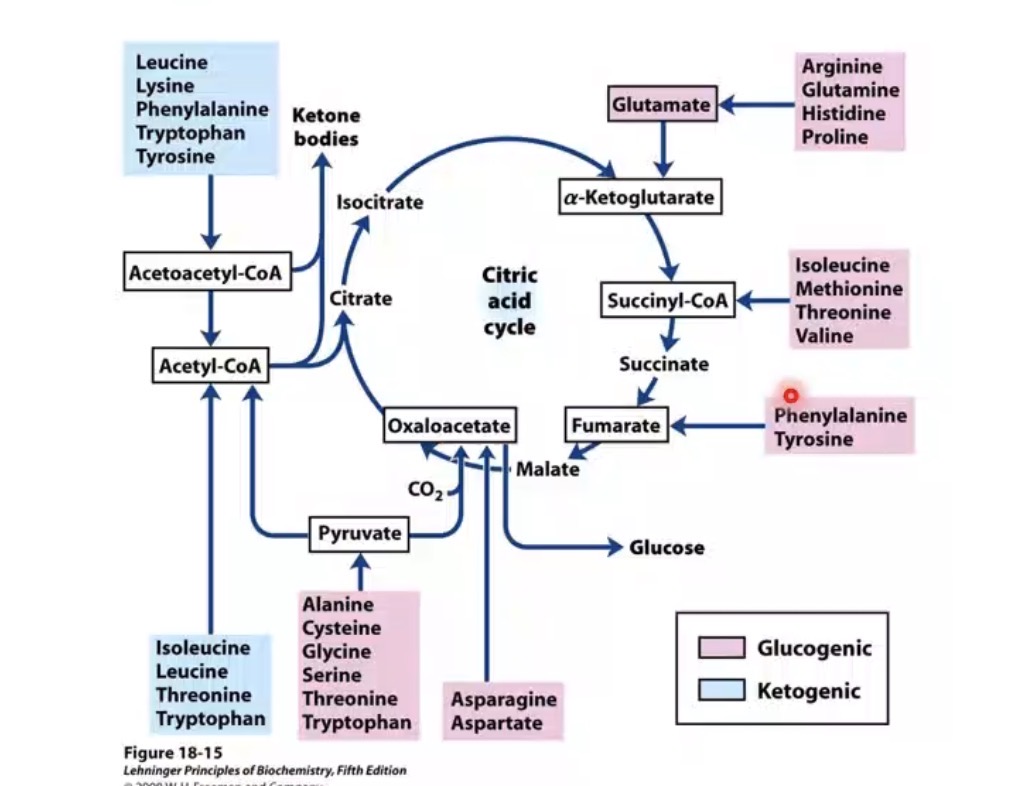
Why are glucogenic amino acids important?
Can support gluconeogenesis or energy production.
What are ketogenic amino acids?
Amino acids yielding acetyl CoA or acetoacetyl CoA.
What do ketogenic amino acids ultimately produce?
Ketone bodies or fatty acid-related intermediates.
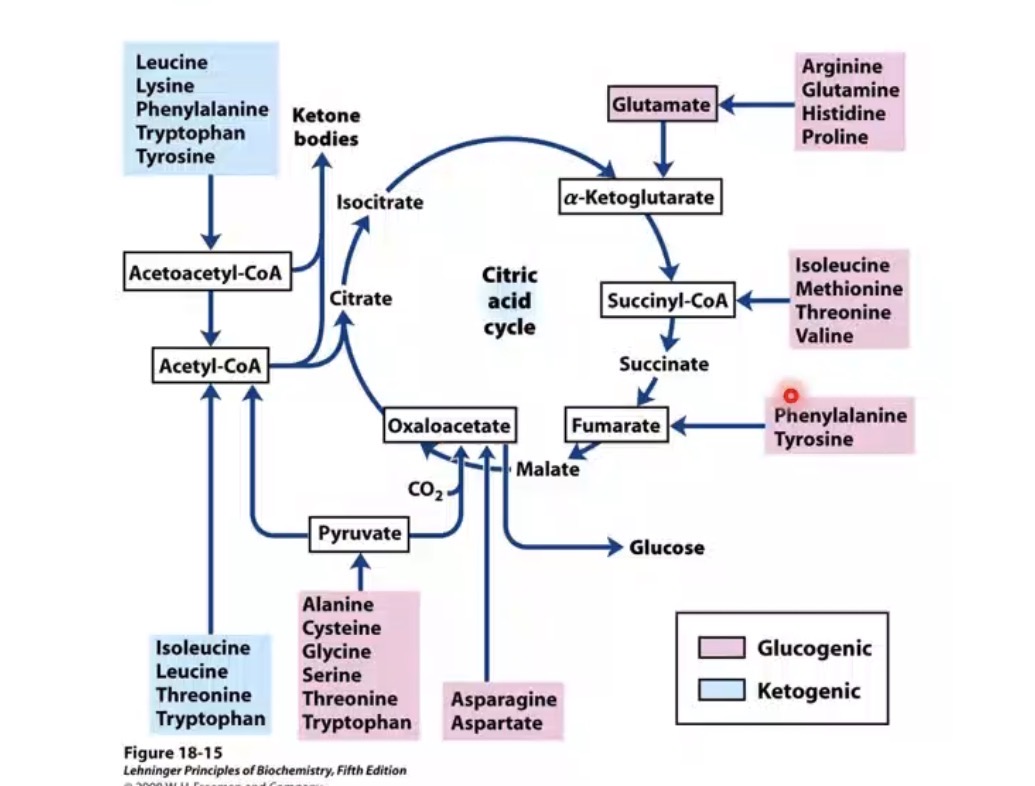
Example ketogenic amino acids mentioned?
Leucine and isoleucine.
Why are branched-chain amino acids important metabolically?
Energy source via complex degradation pathways.
What does acetyl CoA from amino acids do?
Enters energy metabolism.
What happens to glucogenic amino acid carbon skeletons?
Enter the citric acid cycle.
Why are α-ketoglutarate-forming amino acids important?
Feed directly into TCA metabolism.
Can all amino acids eventually contribute to energy production?
Yes.
Why do amino acids provide ATP?
Their carbon skeletons are oxidised.
What central pathway receives amino acid carbon skeletons?
Citric acid (Krebs) cycle.
What is tyrosinaemia?
A disorder caused by impaired tyrosine metabolism.

Cause of tyrosinaemia mentioned?
Tyrosine aminotransferase mutation.

What happens in tyrosinaemia?
Tyrosine accumulation and crystal formation.

Effects of tyrosinaemia?
Eye damage, skin effects, developmental problems.
What is PKU?
Phenylketonuria.
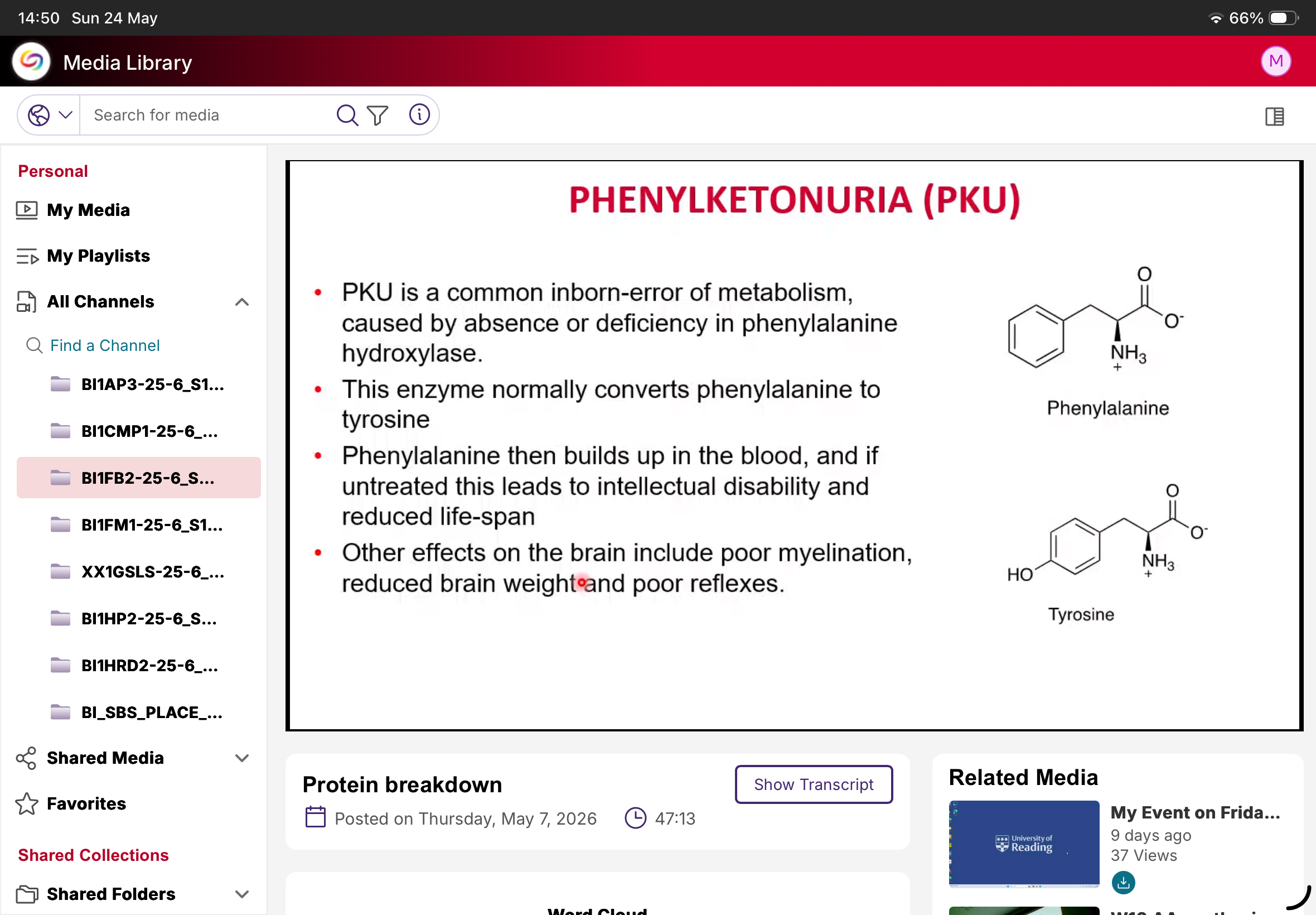
Cause of PKU?
Deficiency/absence of phenylalanine hydroxylase.
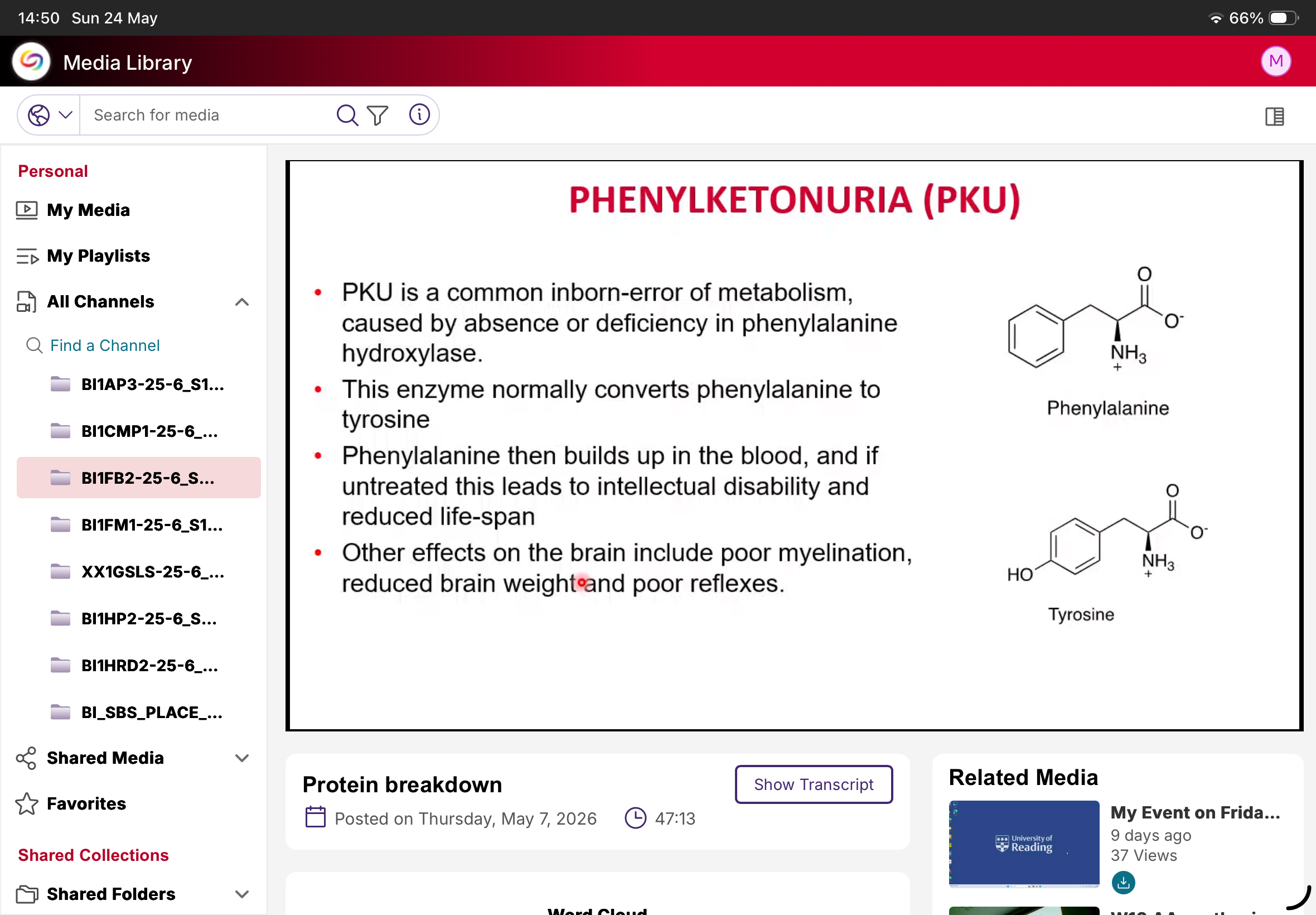
Normal function of phenylalanine hydroxylase?
Converts phenylalanine to tyrosine.
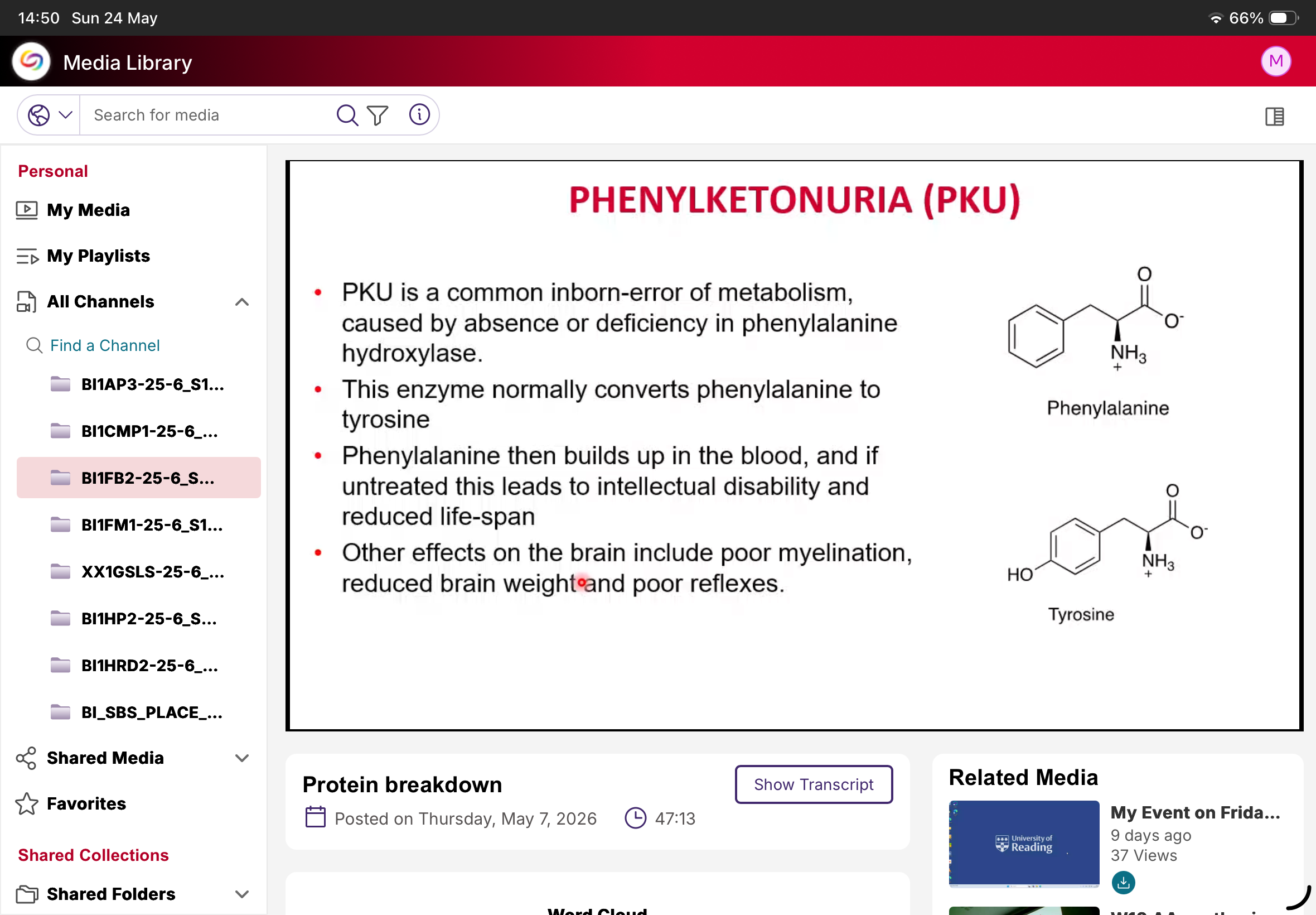
What happens in PKU?
Phenylalanine accumulates.
Why is PKU dangerous?
Causes neurological damage if untreated.
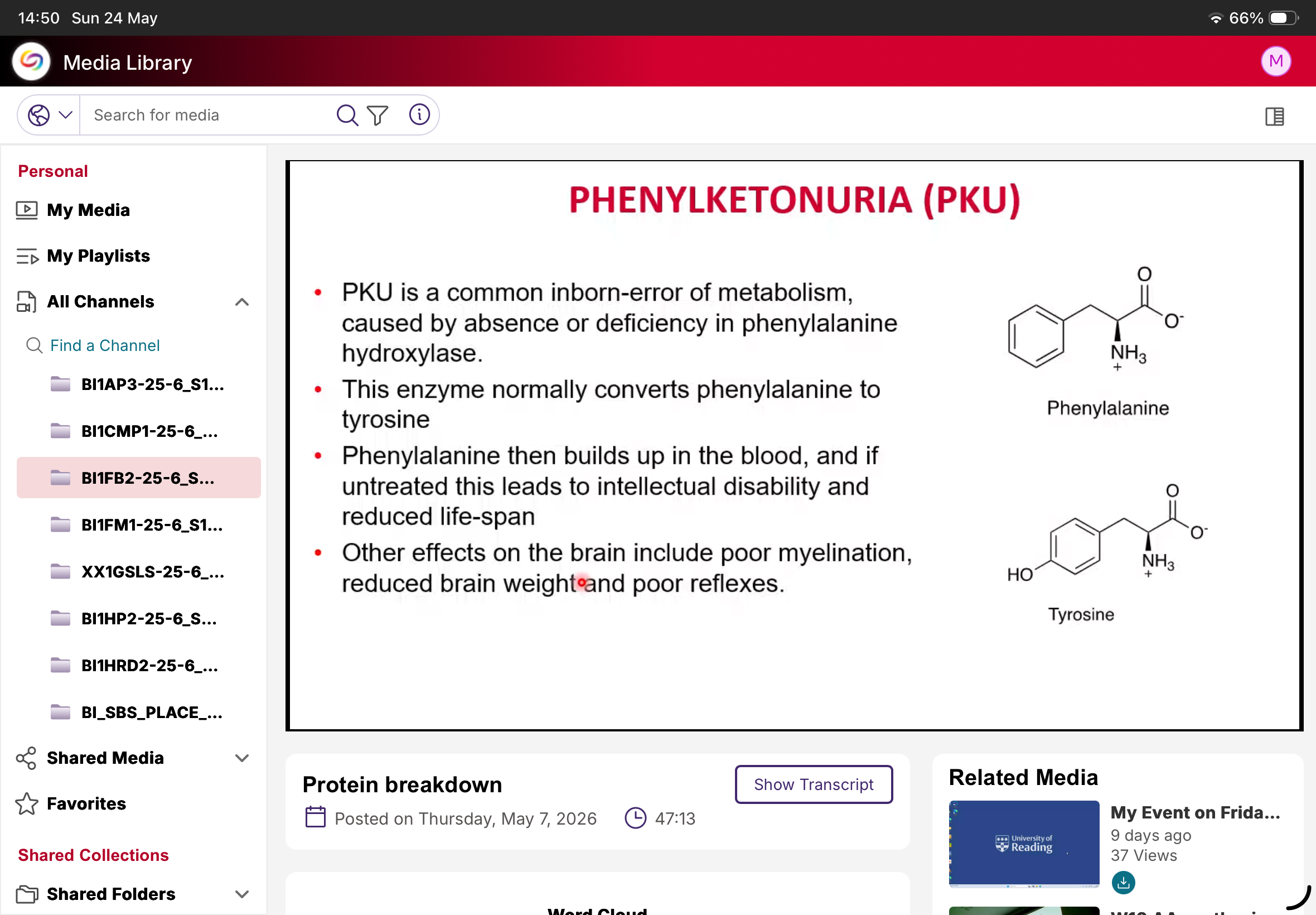
Neurological effects of PKU?
Intellectual disability, poor myelination, reduced brain weight, poor reflexes.
Why does PKU reduce neurotransmitter production?
Low tyrosine and disrupted transport of aromatic amino acids.
Which neurotransmitters are affected in PKU?
Dopamine and noradrenaline.
What is alkaptonuria?
A disorder of phenylalanine/tyrosine breakdown.
Cause of alkaptonuria?
Homogentisic acid oxidase deficiency.
What accumulates in alkaptonuria?
Homogentisic acid.
Why does urine turn black in alkaptonuria?
Oxidised homogentisic acid darkens urine.
Complications of alkaptonuria?
Cartilage damage, heart valve damage, kidney/organ stones.
Why are amino acid metabolism disorders clinically important?
Toxic metabolite accumulation and energy metabolism disruption.
Overall purpose of protein breakdown?
Amino acid recycling, energy production, nitrogen disposal, cellular quality control.