GENE221 Whole course flashcards
1/109
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
110 Terms
What are the five goals of a plasmid evolutionarily?
Replicate – Large plasmids typically only 1-5 copies/cell (low copy number) – Small plasmids ~15-50 copies/cell (high copy number)
Keep host happy – by constraining metabolic load by regulating copy number
Segregate – ensure daughter cells receive at least one copy upon division
Keep host under control – kill off cells that lose the plasmid
Spread – conjugation!; non-conjugative plasmids are often mobilisable
What is the main mechanism for plasmids to keep their replication under control?
– Plasmid DNA replication controlled by plasmidencoded inhibitor that acts at oriV
– As cell size increases, inhibitor concentration decreases and plasmid replication initiated
– Replication results in further copies of inhibitor gene and more inhibitor which limits plasmid replication again
The strength of an inhibitor is proportional to the size of the plasmid, large plasmids will have stronger inhibitors so less of them are replicated, to equal the metabolic load on the bacteria as smaller plasmids which will allow themselves to be replicated more
What is the OriV
Origin of “vegetative” replication
How do high-copy plasmids regulate their replication?
High-copy plasmids have a gene after their OriV on their genome called Rom or Rop that expresses a protein called the Rop protein which stabilises the bonding between an anti-sense RNA called RNA I and RNA II. When free and unbound, RNA II binds to the ColE1 in the OriV to start DNA replication (in E.coli). Thus if the concentration of plasmids and therefore the Rop protein are too high, the RNA II will not be free to initiate plasmid DNA replication.
Fat plasmids don’t want too many of themselves as they are fat chuds so the Ropmaxx to avoid reproduction
How is plasmid replication controlled in low-copy plasmids?
Controlled by the RepA gene, which is adjacent or near the OriV. At high concentration of the plasmid and therefore RepA protein the protein binds to iterons (short, directly repeating sequences of DNA) and binds two identical iterons on different homologous plasmids “handcuffing them together and preventing DNA replication of either of them.
RepA gene (representative of Alabama) handcuffs the little mexican plasmids as they breed to fast and are immigrants in the bacteria, so they grab two iterons and bind them together, handcuffing and arresting the mexican immigrants.
How do high-copy plasmids ensure that they are partitioned so are passed into both daughter cells during bacterial replication?
They don’t do anything actively, there are enough of them that they can be confident statistically that a few plasmids will end up in each cell.
How do low-copy plasmids ensure that they are partitioned so are passed into both daughter cells during bacterial replication?
Controlled by the RepF1A gene in
Why are double auxotrophs used in experiments surrounding horizontal gene transfer of bacteria?
A point mutation causing auxotrophy has about a 1 in 10^6 chance of reverting due to a secondary mutation, during reproduction. This is a high enough frequency to confound with other kinds of reversion or genetic changes due to horizontal gene transfer. Thus a double revertant is used which is auxotrophic for two amino acids, and thus a wild type mutant that could grow on minimal media has a probability of mutation of 1 in 10^12, which is small enough to ignore.
How does a Hfr bacteria form from an F+ bacteria and how does it promote horizontal gene transfer?
An F+ bacteria contains an F plasmid. Through recombination the F plasmid can occasionally integrate itself into a bacterial chromosome. This form an Hfr which is a High-frequency-of-recombination bacteria. This bacteria can still pass on its F plasmid through conjugation to an F- bacteria and often parts of the chromosome are taken with it. From there, another recombination event can cause the chromosomal DNA to be transferred into the chromosome of the other bacteria. This is a common driver of horiztonal gene transfer in bacteria.
What genetic element in bacteria causes conjugation to be inititatied?
The F plasmid, which has the ability to form a Pilus between adjacent bacteria and move through it as a method of spreading itself, as bacteria will replicate it using their own DNA replication systems.
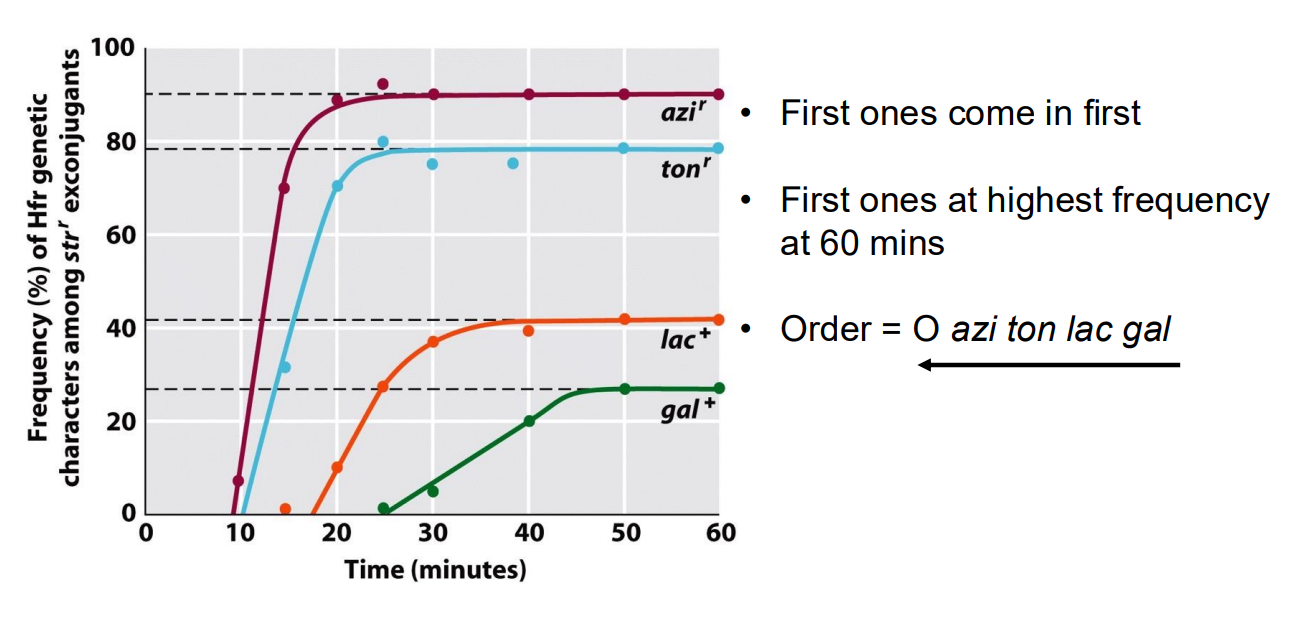
How can we determine the relative position of genes using rate of conjugation of the genes into another bacteria?
The conjugation pilus is quite unstable, so it often breaks while DNA is being transferred. Usually, only a fragment of a chromosome is transferred between bacteria. The F plasmid is always transferred first, so genes directly after the F plasmid in the sequence will be transferred much more often than genes further back in the sequence.
We can mix Hfr Bacteria with mutant auxotrophic F- bacteria that cannot synthesize certain amino acids, thus only bacteria that are subject to conjugation, causing them to be able to grow on media without a supplemented Amino Acid. By the higher frequency of mutants forming on media with different amino acid supplements, we can determine the relative positions of different genes in bacterial chromosomes that code for amino acid synthesis pathways.
How does the F-plasmid save energy by not transferring itself to bacteria that already contain an F-plasmid?
Another function of the F plasmid is that it causes the cell to produce signals on its membrane that indicate to outside bacteria that it already contains an F plasmid, thus another F plasmid won’t waste energy transferring itself.
What is another, more archaic way of measuring relative positions of gene in a bacterial chromosome.
We can artificially cut off the DNA transfer process of conjugation through the pilus by blending
How are F’ factors formed and what are they?
When an integrated F plasmid in a Hfr recombinant bacteria is excised, usually due to crossing over with itself, it can be cut out of the chromosome and reforms into its own circular piece of DNA. Other parts of the chromosome adjacent to the F plasmid could be included in this excision. When entire genes are excised with the F plasmid it forms an F’ plasmid, which has the same virulence as an F-plasmid but can pass on chromosomal genes to F- bacteria. This is another driver of horizontal gene transfer.
What is the DNA transfer carrier in transduction and how does it cause transduction?
Bacteriaphages can accidentally repackage bacterial DNA when forming new phages before cell lysis. They can then transfer the DNA into other bacteria at low frequencies.
What are the limitations/bottles necks of transduction?
Amount of DNA transferred limited by size of phage head – e.g. ~90 kb for P1 (for transduction in E. coli)
Phage host specificity determined at least in part by cell-surface receptors/ transduce genes into same species
In what stage of growth are bacteria receptive to transformation and how is this triggered?
Bacteria use quorum sensing to determine the density of homologous bacteria in its area, so that when the density is sufficiently high, the bacteria goes into a state called competence. This is when a bacteria can absorb free DNA in solution and incorporate it into its own.
What other event is accompanied with high density of bacteria and competence induction?
The cell lysis of 5-20% of the bacteria in a group, causing their DNA to release into solution to be picked up by bacteria in a competent state. This increases genetic diversity/in the population at a small cost of less total population and is a main way that bacteria can pass on antibiotic resistance genes.
What is an auxotroph?
a micro-organism that can only grow in the presence of specific growth supplements such as amino acids
In the one gene-one enzyme experiment carried out by Beadle and Tatum, what steps were taken to find loci (genes) in which mutations caused arginine auxotrophy?
They placed a mutant of an organism (Neospora), and determined that it was auxotrophic for arginine by trying to cultivate 20 colonies of Neospora on minimal media + 1 amino acid. The only colony that grew was in minimal media +arginine, indicating the mutant was arginine auxotrophic.
They carried out a detailed study of compounds that would enable the auxotrophs to grow, identifying three gene mutations that caused loss of function of the synthesis of three different compounds in the arginine synthesis pathway, thus having three classes of mutants: arg-1, arg-2, and arg-3.
In the one gene-one enzyme experiment carried out by Beadle and Tatum, what steps were taken to determine what different mutants arg-1, arg-2 and arg-3 caused?
The three mutants were exposed to minimal media + three different compounds, either arginine or one of its precursors in the biosynthesis pathway, and the following results were found.
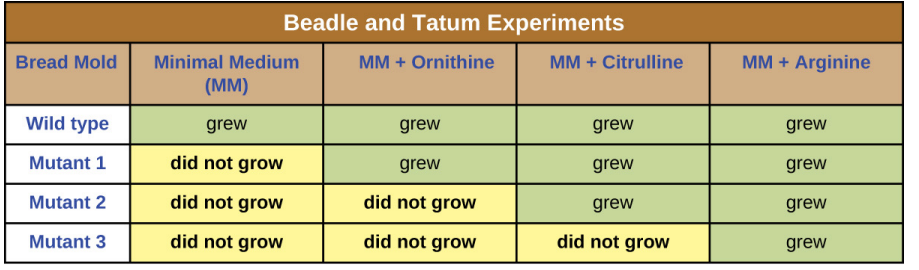
In the one gene-one enzyme experiment carried out by Beadle and Tatum, what did they conclude from their results of growing arg-1, arg-2 and arg-3 mutants in different media?
Each gene arg-1, arg-2 and arg-3 gives rise to a different enzyme that catalyses a different chemical reaction.
This formed the basis of the one-gene-one-enzyme hypothesis: “each gene in an organism corresponds to a single enzyme, and vice-versa ”
how did the fluctuation test show that mutations arise spontaneously in a population, and are not due to the presence of phage inducing some of the bacteria to become resistant (Induced mutation).
If induced mutation was the cause of antibiotic resistance, we would expect resistance to arise naturally within all or many different unique bacterial environments at a reasonably similar rate. If resistance arose spontaneously in a population, it would be a genetic mutation that would be passed on and as such, resistant bacteria may explode in population. This being a small chance, some cultures would have no bacteria, and some would have lots, thus there would be a large fluctuation in bacteria colonies that grow in a given culture.
Upon performing the test, a massive fluctuation in bacteria colony numbers was found, and so spontaneous mutation was ruled as the mechanism of mutation in organisms.
Define Mutagen
an agent that is capable of increasing the mutation rate
How does the Ames test allow detection of mutagens
uses strains of a bacterium Salmonella typhimurium that are AUXOTROPHS - have a mutation in a gene required for synthesis of histidine, an amino acid (His- phenotype; hisC or hisG genes)
• Bacteria are spread onto a growth medium that does not contain histidine
• Any bacteria that can grow have had a second mutation and are revertants - have reverted (gone back) to being able to make Histidine
• If the test compound increases the number of revertants it is likely to be a mutagen
How was the Ames test improved to make it more appropriate for testing mutegenicity on mammals?
many chemicals that are not mutagenic to bacteria are mutagenic in mammals, because these chemicals are converted into an active form in mammals by enzymes in the liver, after they have been ingested
• solution: prepare liver extracts and incorporate them into the medium used in the Ames test
What are some ways to increase to rate of mutation in the Ames test generally?
Added features of the bacterial test strains:
(i) Mutation in a gene (rfa) that affects the cell envelope - makes the bacteria more permeable to some chemicals
(ii) Defective in a gene (uvrB) encoding a protein that repairs damaged DNA and reduces the frequency of mutations
(iii) Contain a plasmid (pKM101) that enhances the effectiveness of some mutagens
ALL OF THESE INCREASE THE EFFECTIVENESS OF MUTAGENS IN CAUSING MUTATIONS
What are three common types of mutations?
1. point mutations = mutations at a single point (!) (most often a single base pair) in a genome – SNPs (Single Nucleotide Polymorphisms)
2. can be substitution mutations or indels (insertion or deletion of base-pairs)
3. can also be deletion (loss) or large-scale rearrangements of DNA or insertion of mobile genetic elements
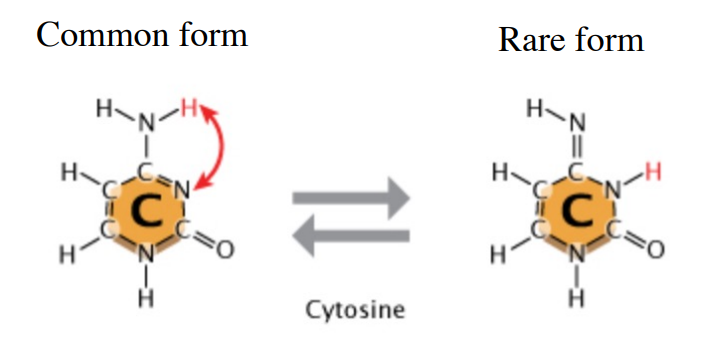
How does tautomerisation affect DNA replication/cause mutations?
Tautomerisation can occur, where a cytosine molecule switches to a rare tautomer. This different kind of cytosine bonds to Adenine instead of Guanine. (Goes from 3 H-bonds to 2). This causes a GC > AT mutation.
How are DNA point mutations fixed during or after DNA replication?
Both DNA pol I and DNA pol III serve a “proofreading” function by excising incorrectly inserted mismatched bases.
Once the mismatched base is removed, the polymerase has another chance to add the correct complementary base.”
What is another mechanism that fixes point mutations in DNA
Mismatch repair (MMR).
The first step in MMR is the detection of mismatches in newly replicated DNA by the MutS protein.
Binding of MutS to distortions in the DNA double helix recruits MutL and MutH.
MutH cuts the newly synthesized strand containing the incorrect base.
Without the ability to discriminate between the parental and newly synthesised strands, the MMR system could not determine which base to excise.
How does the MMR system differentiate between the original and new strand in E.coli?
DNA inside E. coli is chemically modified by methylation of adenine bases in the DNA.
Strand recognition by MutH is directed by adenine methylation at GATC sequences.
Because adenine methylation occurs after DNA synthesis, newly synthesized DNA is temporarily unmodified, and this temporary absence of methylation directs repair to the new strand.
The MutH endonuclease cuts the unmethylated strand
What is a random chemical event that occurs on cytosine, causing a GC > TA mutation?
Deamination of Cytosine.
Deamination = the hydrolytic removal of an amine group
Deamination converts cytosine to uracil. Uracil base pairs with adenine in replication, converting a C · G base pair into a T · A base pair.
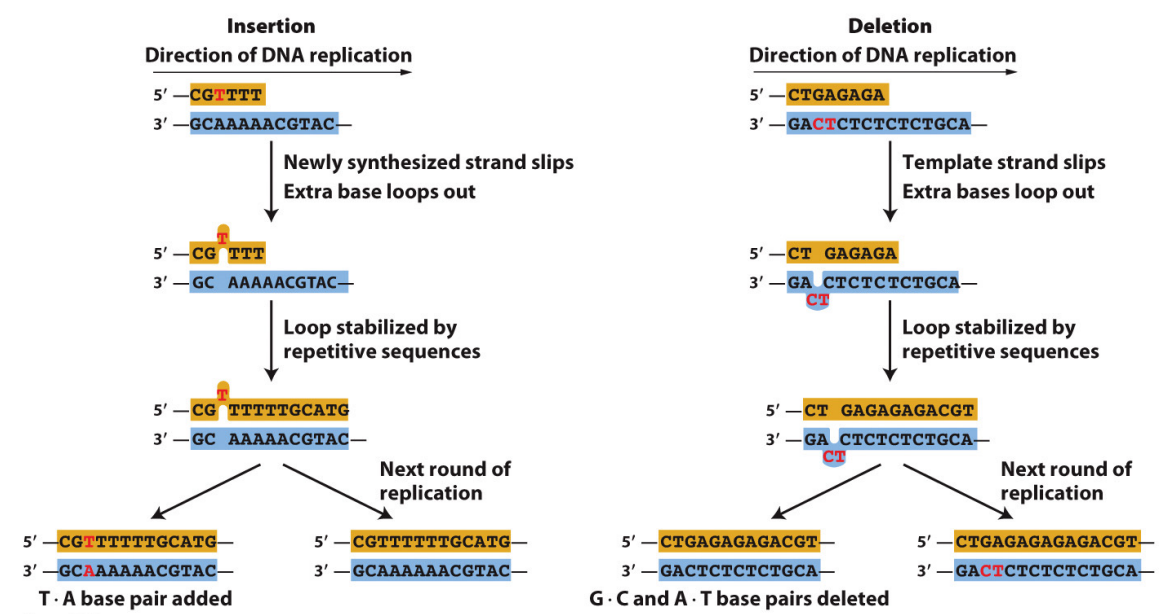
What are the two main ways that InDel mutations can occur?
Base insertions and deletions (indels) are also caused by DNA replication errors.
Indels arise when loops in single-stranded regions of DNA are stabilized by the “slipped mispairing” of repeated sequences in the course of DNA replication.
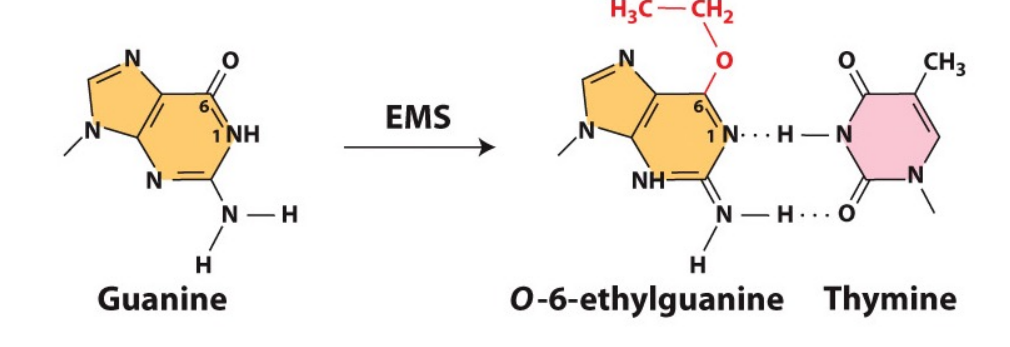
How does the chemical mutagen EMS (ethyl methane sulphonate) cause mutations? And how are these potential mutations corrected?
EMS is an alkylating agent that changes Guanines configuration to bond with Thymine, causing GC → AT mutations.
Repair proteins – alkyltransferases. The (alkyl) ethyl group is transferred from the base in the DNA to the protein
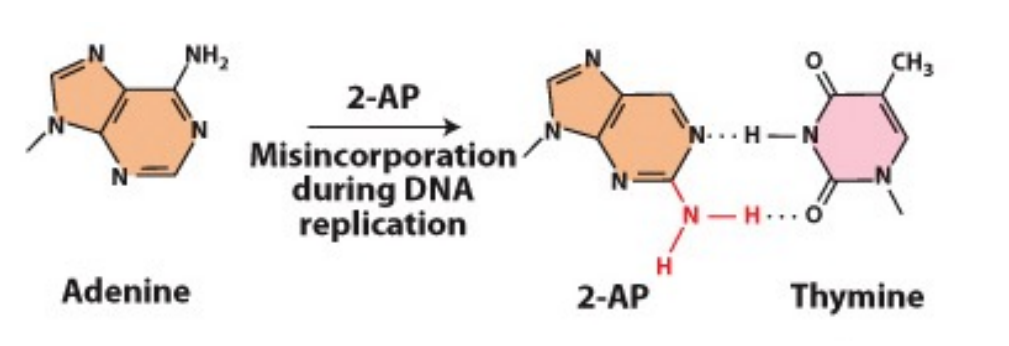
How does 2-AP (2-amino purine) cause mutations?
- Most often pairs with thymine but can also base-pair with cytosine
- If this happens during replication, will lead to an A-T basepair being changed to A-C and subsequently, G-C
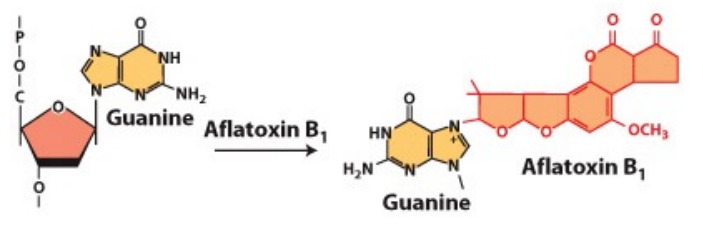
How does Aflatoxin cause mutations?
•produced by fungi
•chemically reacts with guanine (G) bases in DNA, generating apurinic sites. This can lead to mutation
•causes GC →TA mutations
What are apurinic sites and why do they cause mutations?
• Depurination - a purine base (adenine or guanine) is lost from the DNA
• This gives an apurinic site, ie. a site without a purine
• during DNA replication, there is a "blank" where the purine should be
• a base (often an adenine) may be inserted opposite the blank
• this can change the sequence of base-pairs
How does radiation cause mutation?
When hit by a high-energy UV ray, adjacent thymidine (T) bases in the DNA can become covalently crosslinked - photodimers •
These fail to base-pair properly during DNA replication
• Translesion DNA polymerases (bypass DNA pol) can replicate past these but may incorporate the wrong base as they have significantly less error-correction
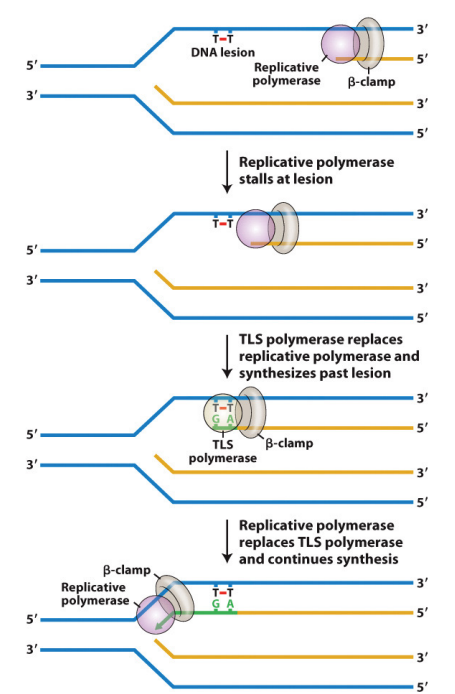
How does Trans-lesion DNA synthesis occur?
It is initiated by stalled DNA polymerase, which triggers the recruitment of a TLS polymerase that synthesizes past the lesion.
Once extension passes the lesion, the TLS polymerase is replaced by the replicative DNA polymerase.”
Conserved from E. coli to humans.
What is a pre-mutagenic lesion and how do they effect mutation?
a change to the DNA that may lead to a mutation.
• At least one round of DNA replication is needed to get from a pre-mutagenic lesion to a mutation
• DNA repair takes place at the pre-mutagenic lesion - once the mutation is "established", it is too late
• DNA repair systems repair “damaged” DNA BEFORE it is replicated
How are apurinic sites in DNA error-corrected?
An enzyme (AP endonuclease) recognises an apurinic site and cuts the strand of DNA that contains it. The defective DNA and some adjacent DNA is then removed by a set of enzymes (excision exonucleases). The gap is filled in by DNA synthesis.
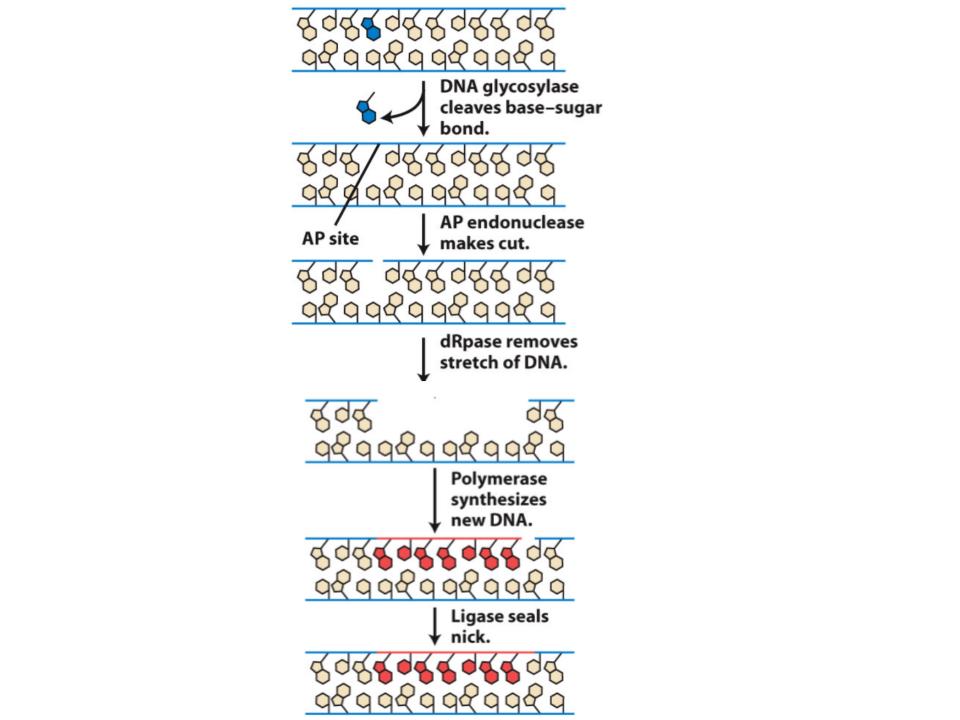
How are photodimers caused by radiation error-corrected so as not to cause mutations?
Multiple mechanisms are known e.g
Nucleotide excision repair similar to repair of apurinic sites (Fig. 15-16 in Griffiths et al 12th ed).
Photolyase enzyme - this uses energy from white light to convert photodimers back to pyrimidines (photorepair)
Define Complementation
production of wild-type phenotype when two mutant haploid genomes bearing different recessive mutations are present in the same cell
Briefly describe how Benzer’s complementation experiment discovered complementation
Benzer infected colonies of E.coli bacteria with two different mutant bacteriaphages that had mutations making it unable to infect this strain of E.coli. Occasionally, the phages would be able to infect E.coli, which was observable as holes in the E.coli colony growth on a petri dish. This is due to complementation, as phages share functional genes from their DNA with mutant phages with non-functional genes, so that they can both infect cells and lyse. This sharing of DNA is not passed onto progeny, so both bacteriaphages produce progeny identical to the parent.
If two mutant bacteriaphages cannot perform complementation with each other, what does this mean for the mutation/s they have?
The mutations are on the same gene, so neither bacteriaphage has a functional copy of the gene to share.
Define Recombination
a process that generates new gene combinations
How is recombination used in Benzer’s experiment to make bacteriaphages that can infect E.coli strain K
Using two different rII mutant bacteriaphages to infect E.coli strain B which the phages can infect. During this infection, very rarely the chromosomes of the bacteriaphages can cross over and occasionally produce wild-type phages that lack the loss-of-function mutations for those genes, making them able to infect E.coli strain K.
Describe the basic steps of making a plasmid clone in E. coli
1. Cut a plasmid (vector) and DNA to be cloned with same restriction enzyme
2. Use DNA ligase to insert DNA into plasmid
3. Transform into E. coli
4. Screen for plasmids carrying insert of interest
What is blue-white selection and why is it used?
Blue-white selection is a common method to select for bacteria containing recombinant DNA. We do this with a vector containing an Ampicillin-resistance and a lacZ gene, like the pUC vector, as well the gene of interest to be transformed into the bacteria. We can plate out colonies of potential transformed bacteria onto a solution of Ampicillin (to control for untransformed bacteria) and X-Gal. The pUC vector has a MCS in the lacZ gene, so a restriction enzyme will make a cut there and the gene of interest could ligate into this site, bisecting the lacZ gene and making it non-functional. This means the bacteria cannot cleave X-Gal, so transformed bacteria with the insert of interest will show up as white, while transformed bacteria without the insert will be blue.
Explain how gateway cloning works
Gateway cloning doesn’t use restriction enzymes and DNA ligases, instead it takes advantage of a site-specific recombination system from the Lambda phage. The target gene of interest that lies between two Phage attachment points can be recombined onto a different Destination vector (which contains a selectable marker like AmpR, replacing the gene that was between two corresponding phage attachment sites. This makes a final Expression vector containing AmpR and the target gene insert. This plasmid can be transformed into many different organisms so is a very versatile tool.
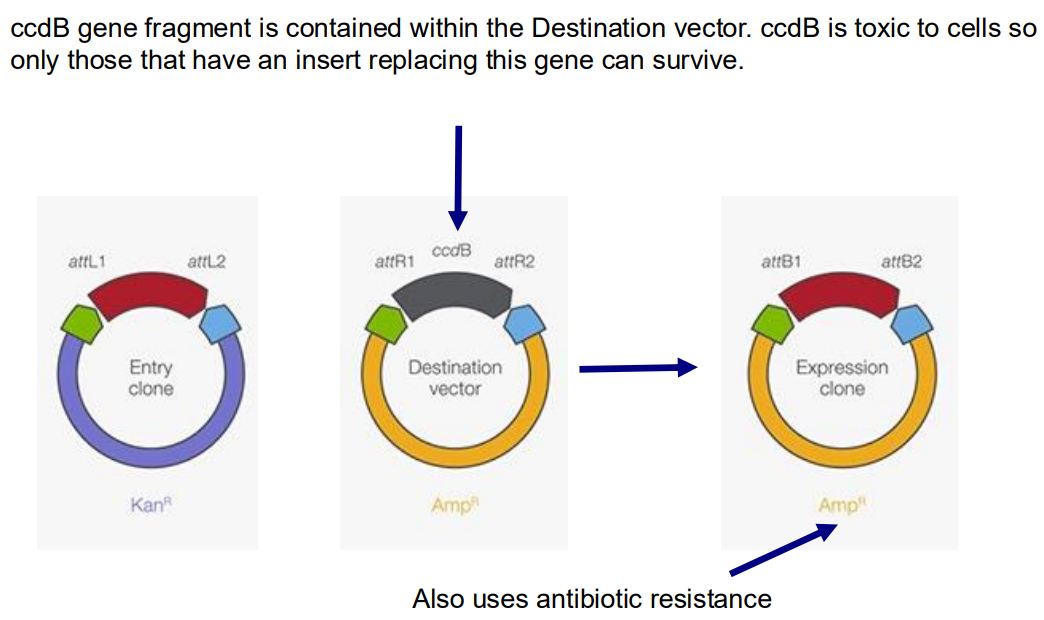
Why do we make expression vectors, and what techniques are used to alter their function?
Expression vectors are used to produce protein products in high amounts. We can control expression by using a Ptac promoter which is induced by IPTG, we can also use stronger promoters with higher binding affinities to increase the rate of expression and therefore protein production.
What techniques are used to purify the protein product from an expression vector solution?
We can add a magnetic or electrically charged tag to the protein-coding sequence of the protein of interest, with a thrombin cleavage site between them. Then we lyse the cell solution and pass it through a column, the tag on the protein will stick to the column. Then we can mix the stuck tagged protein solution with thrombin which is a highly specific protease. This removes the tag and gives us a purified protein product.
How can GFP proteins be used to measure gene expression?
We could use Restriction Enzymes to digest different fluorophores, Ligate this DNA into different plasmids (with different promoters) to generate fluorescent plasmids. Transforming these different fluorescent plasmids into bacteria, we could then compare the colour expression of different fluorophores and different promoters.
What is another expression protein that we can use to quantitatively measure gene expression?
The Luciferase gene produces the luciferase enzyme. We can clone a promoter upstream of this gene, in a vector, and measure the emission of the light produced by the gene with photospectroscopy to find the extent of the promoter’s transcriptional efficiency.
Outline TA cloning to clone PCR products
Taq DNA polymerase adds on an adenine at 3’ end of PCR product. We can take advantage of this by treating vectors with restriction enzyme that leaves a thymine, T, overhang on both sides. The vector can’t religate on itself, so only clones with inserted DNA (from the PCR product) will survive.
What DNA cloning method is used to join multiple DNA fragments together?
Gibson Assembly. This is a method to join around 10 DNA fragments together. The DNA fragments must have overlapping/identical ends so they can bind and be filled in with DNA ligase and polymerase and then cloned with PCR, this will form a continuous strand consisting of all ten DNA fragments. This is done with three main enzymes in a cocktail, T5 endonuclease, DNA ligase and DNA polymerase.
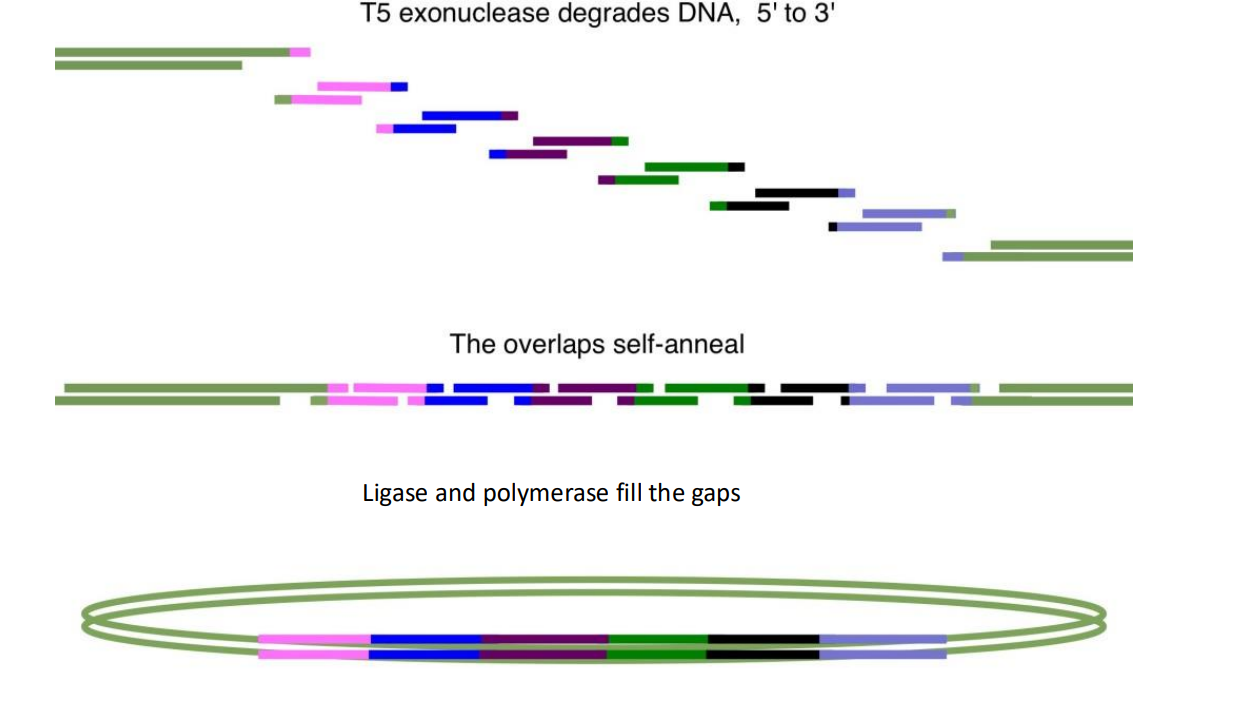
In what situation would you use synthetic DNA made in a lab (gblocks)
If you needed a specific DNA sequence and it was difficult to PCR or isolate
if the gene sequence is not found in nature
To save time and potentially money if cloning the DNA required exotic materials.
What is a genomic library?
A collection of all the genomic DNA fragments of a given species that have been taken from one organism and inserted into a type of vector for cloning.
What is a cDNA library?
This is a library without the introns added into the genome. This is made by taking mature RNA from an isolated cell or tissue and using reverse transcriptase to turn it back into DNA, but the introns were removed when turning it to RNA so it make cDNA, complementary DNA that i sonly the coding portions.
Describe the process of Genomic Shotgun sequencing
You obtain many copies of a genome from an isolated cell or tissue. You then break up the sequences into smaller DNA fragments using sonic waves or enzymatic lysis. You can then clone these fragments onto vectors to make a gDNA library. Then you can sequence the DNA on these vectors and overlapping sequences will allow you to piece together the sequence of nucleotides in the genome.
Define Lytic
the phage lifecycle that results in lysis of the bacterial cell upon release of progeny phage
Define Lysogenic
the phage lifecycle that results in stable carriage of the phage (prophage) within the host cell (lysogen)
Define Virulent Phage
A phage that is only able to undergo replication via the lytic cycle
Temperate Phage
A phage that can replicate via either the lytic or lysogenic cycles (e.g. phage lambda)
Define Lysogen
a host cell that is harbouring a prophage during lysogeny
Define Prophage
the latent form of a temperate phage that remains within the lysogen (e.g. integrated into host chromosome)
What are the three DNA-binding proteins in the Lambda phage?
Cro is a DNA-binding protein that represses transcription – Cro promotes the lytic cycle
CI is a DNA-binding protein that can activate or repress transcription – CI activates it’s own expression – Represses genes required for lytic cycle – Maintains lysogeny
CII is a DNA-binding protein that activates transcription – CII promotes the lysogenic cycle
Cro(0) represses, CI(1) in the middle, CII(2) activates.
How do phages know when to break enter to lytic or lysogenic stage?
Host proteases degrade CII Healthy cells produce high levels of protease In actively growing cells, CII gets degraded Cro protein wins the battle and the lytic cycle occurs. If the cells are starved then less proteases will be produced, so CII is produced and the lysogenic pathway goes through.
How does expression of the cro gene cause the lytic pathway to proceed in lambda phages?
The cro gene produces the Cro protein which represses transcription of the CI gene and most genes that were expressed before the cro gene. The Cro gene activates genes for the lytic pathway, constructing phage heads and tails for example.
Why is it that while the lysogenic cycle is being maintained in a bacteria, that bacteria is imune to bacteriaphages of the same kind?
While the lysogenic cycle is being maintained, so a bacteriaphage genome is integrated into a bacterial chromosome, the CI gene in Lambda phages acts as a repressor for any lytic genes, including those in other lambda phages in a bacteria. So infecting bacteriaphages have their lytic pathway repressed by the CI gene in the lysogenic phage.
How does the lysogenic maintenance gene CI regulate its own expression
There are three main operator sites that the CI protein can bind to, OR2 and OR1. Binding to these sites blocks the cro protein from being produced, which would start the lytic pathway. OR3 has a much lower binding affinity to CI and is upstream of the CI protein itself. If the concentration of CI protein in the cell becomes too high, then CI will be able to bind to OR3, which acts as a repressor, so it blocks RNAP from binding to the promoter and causing more expression of the CI gene, thus regulating its concentration in the cell.
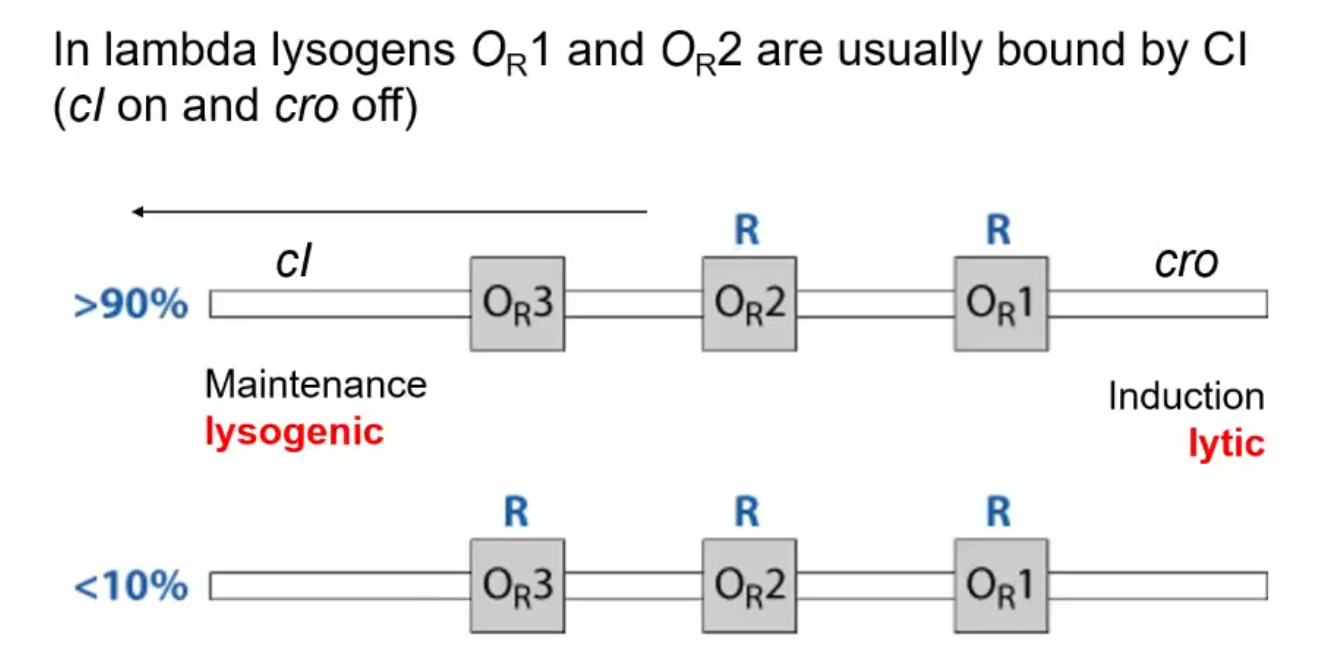
How is the CI protein an activator of itself?
Cooperativity: Bound CI at OR1 increases affinity for OR2 (aided by protein-protein interactions).
How does the lambda phage switch from being in the lysogenic cycle to the lytic cycle?
Induction via UV results in DNA damage that is somehow sensed by RecA resulting in it becoming a co-protease. Activated RecA assists CI cleavage into two domains (nonfunctional) and therefore clears the operator sites of CI. This allows for the cro gene to be expressed, and the Cro protein activates the genes required for the lytic pathway.
Describe the three-domain tree of life and explain why molecular data changed our view of microbial diversity.
The three domains of life are bacteria, archea and eukarya. Historically the phylogenetic tree was thought of as dominated by eukarya but this was due to sampling bias. More recently, the prokarya branch has been split into bacteria and archaea. Archaea are also single-celled organisms but are difficult to grow and isolate in culture so only recent molecular data has illuminated our perception of them. They used to be thought of as extremophiles but recently have been found in more balmy environments like human guts.
Explain how ribosomal RNAs and conserved proteins can be used to infer evolutionary relationships.
Ribosomes are universal & conserved (essential DNA for translation). Their RNA & protein components especially useful for deep evolutionary comparisons. Hug et al. formed the first mostly complete phylogenetic tree by aligning 16 RNA proteins/genes that are essentially constant across all life and aligned all the samples they could get, then measured them by genetic differences to build a phylogenetic tree.
Define and distinguish the molecular clock, genetic drift, and the nearly neutral theory of molecular evolution.
The molecular clock: the number of mutations within biomolecules can be used to deduce when two or more species diverged i.e. closely related species have similar genomes (DNA, RNA & protein) sequences, more diverged species have dissimilar sequences
The nearly neutral theory of molecular evolution: Most genetic variation between species is either neutral or slightly deleterious and is fixed in the population due to genetic drift i.e. individuals in a population with and without a neutral variant will have almost no difference in fitness
Define homology and distinguish it from similarity and analogy.
A homologous trait is any characteristic of organisms that is derived from a common ancestor Eg. vertebrate forelimbs, or the coronaviruses. Analogy and similarity are commonalities in genotype or phenotype in organisms, usually do to convergent selection pressures causing convergent traits.
Explain why most observed variation in homologous protein-coding genes is neutral or nearly neutral.
Genes that are homologous across distantly related organisms are almost definitely essiential genes. Variation that are deleterious is removed from the population by negative selection (also called “purifying selection”). As a consequence of this sequence conservation over long time periods implies strong negative selection
What are some examples of common nucleotide or amino acid changes that point toward the nearly neutral mutation hypothesis.
The most common kind of point mutation are ones that code for the same amino acid, as they have no effect on protein formation.
Non-coding DNA and RNA are much more likely to have mutations in them.
Indel mutations are mostly likely to be prevalent if they preserve frame (i.e are in multiples of three so do not cause a frame shift in the protein.)
What are the Dayhoff Classes?
Dayhoff classes are amino acids that are grouped together as they f=have functionally similar chemical proteperties that make them likely mutations of each other as they mainly make nearly neutral mutations.
Big-dick DEQN went to a STAGP(arty), chatted up a big titty MILV and went home WYF HRK. C(hud) got no play and is always alone
Recognize how evolutionary selection applies to RNA genes.
Nearly neutral evolution is highly preferred in RNA. RNA structure is conserved over RNA sequence, so nucleotides that are far away in the sequence could be changed in a way that still allows it to base-pair to another nucleotide when the RNA molecule folds up.
Explain why most observed variation in homologous ncRNA genes is neutral or nearly neutral.
There is a strong negative selection for changes in ncRNA since their structure often has to stay similar to be effective at its task. This means the structure of an ncRNA has to be conserved across mutations for that mutation to still be viable for the organism.
Explain the basic design of a genome-wide association study and how GWAS links genotypes to phenotypes.
A successful GWAS requires:
A clear, measurable phenotype (e.g. height, antibiotic resistance, virulence, etc.)
An adequate sample size
Sequencing and SNP calling
Corrected for population-structure &/or phylogenetic-relationships
You have to be wary of: Recombination (e.g. HGT) can help shuffle genomes, linkage can cause false positives.
Explain why statistically significant associations may be misleading because of linkage, population structure, recombination, biased sampling, or multiple testing.
Linkage: This is when genes are physically close to each other so usually stay together through recombination and HGT. This means groups of genes are usually found together, so a whole group of genes may be high associated with a phenotype, but it is likely just one gene that causes it.
Population Structure: Phylogenetic makeup of a population is important to factor in, relations between individuals, ethnic sub-group makeup.
Recombination: Makes phylogenetic tree mapping much more difficult, but can break linkages between genes so good and bad.
Biased sampling: A control group may often come from a non-diverse set or a sub-group of the population. In humans, most genetic testing has been done on rich europeans so comparison with a control group is not necessarily population-representative.
Multiple testing: When testing many relationships, predictors, from the same dataset, eventually one is likely to come up as significant when it is not. Solution is to not think P>0.05 significant, take it heuristically.
How does sample size influence GWAS power and interpretation?
Sample size increases predictive power of GWAS, meaning its ability to detect weak signals, or find predictors/variables to be significant.
How does recombination influence GWAS power and interpretation?
Recombination confounds phylogeny, but breaks up linkage blocks, which gets around the main problem of GWAS. Recombination hotspots may coincide with genes involving responses to selection pressure (e.g. antibiotic resistance).
How does phylogeny influence GWAS power and interpretation
Complex phylogeny tables reflect that extensive recombination has occured in a population.
Why could significant associations of genes with a phenotype in GWAS reflect causation, linkage, or population structure?
The significant association between genes and a phenotype in GWAS could be due to causation if the gene involved did cause in the phenotype being measured. If there is a group of genes all associated at a similar level and near each other in the genome then it is likely that only one or two of the genes acutally cause the phenotype and the others are nearby those causative genes on the genome so move with them during recombination, this is linkage.
Significant association with genes and phenotypes may be due to sub-groups in the population being associated with the phenotype being measured, so characteristic genes of these sub-groups may be overrepresented in GWAS.
Distinguish the causal-effect and selected-effect definitions of function?
Causal effect: for a gene to have a function, it is necessary & sufficient for the gene to be linked to an activity.
Selected effect: for a gene to have a function, 1. the gene is linked to an activity AND 2. the gene and the activity is maintained by selection (i.e. has a fitness effect).
What bioinformatic tests could you perform to link a gene with a function?
Gene knock-outs, fitness, gene expression or interaction assays can be used to link genes with functions.
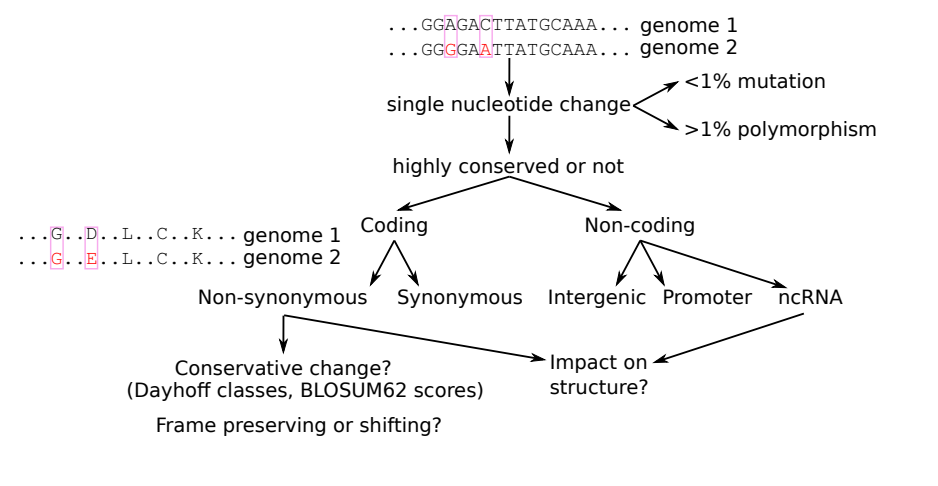
How would we go about classifying variants as non-coding or coding and identify features that may tell us about its effect on the gene function?
Is your variant in a protein-coding region?
Synonymous or non-synonymous (different bp’s, same aa)? If non-synonymous, are they biochemically similar (in the same Dayhoff class)? Is the variant a frame-shift &/or does it introduce a premature stop codon?
Is it a non-coding RNA or not?
Does the mutation preserve or break an otherwise conserved secondary structure? Recall the canonical basepairs: G:C, A:U, G:U. A change from one of these that still resulted in a canonical basepair would likely be conserved in a ncRNA gene.
What are the benefits of using a metagenomics approach over 16S sequencing?
16S sequencing can give us an understanding of the taxes of microbes in a sample and populations within a sample, since it is an RNA sequence that is always present in bacteria/archaea. 18S sequencing is similar but with a distinctive sequence found in all fungi.
Metagenomics can give us far more data about the metabolic potential of a population and their genetic makeup i.e. how they relate to other organisms genetically.
What other benefits can metatranscriptomics offer compared to metagenomics?
Metatranscriptomics is done via Total RNA Sequencing (RNASeq). It can give the same information at genome sequencing but with other advantages.
Advantages:
We can determine gene expression of microbes within natural environments. We can see the diversity of active genes in a community, quantify gene expression under different conditions.
It can provide information about differences in the active functions of microbial communities which appear to be the same in terms of microbe composition
Can be used to study the virosphere! (but also all the parasites, bacteria, fungi = infectome) as well as the host gene expression
What types of analysis can you do with metatranscriptome data?
De novo assembly: putting short sequence reads together to reconstruct longer sequences (contigs). Contigs = contiguous sequences from shorter, overlapping reads. Annotating: once assembled, identifying where the contigs came from using sequence homology
Map sequence reads to a reference sequence
What causes for bias does metagenomics/metatranscriptomics have?
There is potential for sampling bias in data collection. The following points are important to think about.
Sequencing platforms – error rate, biases, read length, noise
• Coverage/depth
• Sample collection and preservation - contamination can overwhelm the real signal
DNA/RNA extraction methods
Nucleic acid extraction from all cell types to avoid bias
Should be effective for diverse microbial taxa
Very small quantities are usually sufficient for sequencing
• Enrichment or depletion steps can affect microbes.
• One sample is only representative of a single time point
What limitations does metagenomics/metatranscriptomics have?
Many genetic sequences are left not annotated because we don’t know what protein they encode. Our understanding of microbial communities is partial based on what we can infer from existing knowledge (i.e. what is well-characterised and exists in databases). We need better computational tools to deal with all the data we can now produce, which is where AI comes in.
Dark matter in metatranscriptomics makes up a vast portion of the transcript data we produce, but transcripts with no sequence homology to anything we understand so are left unannotated.
• What can artificial intelligence offer to metatranscriptomics?
The metagenomic identification of viruses is currently limited to those with sequence similarity to known viruses. Highly divergent viruses that comprise the “dark matter” of the virosphere remain challenging to detect. Over the past decade, artificial intelligence related approaches, especially deep learning algorithms (i.e. AlphaFold), have had a huge impact on protein structure prediction. Possibly AI could be used to help uncover more “dark matter”
What factors seemingly shape the composition of the viral communities?
Virus emergence is often caused by the disruption or invasion of an ecological niche.
Abiotic and biotic factors have been identified that potentially modulate microbial community diversity and structure.
Identifying factors that promote diversification is crucial to understanding disease and its emergence. For example, host ecology and behavior affect contact rates among hosts, and are therefore likely to be important in shaping viral dynamics
What is preferential host switching and why is it important for understanding virus virulence and emergence?
Viruses are more easily transmitted between closely related hosts. If exhibited in real-world conditions, preferential host switching would mean that host taxonomy plays a key role in shaping virome composition.