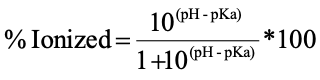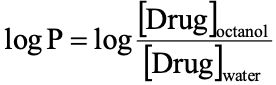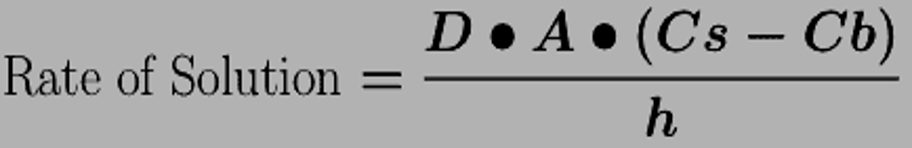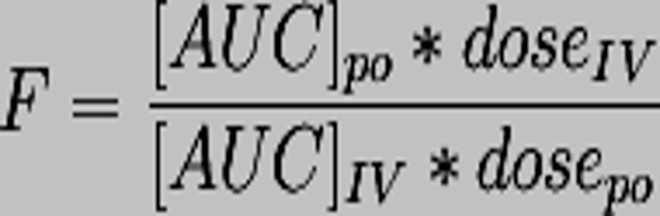Looks like no one added any tags here yet for you.
Biopharmaceutics
study of effect of physical and chemical properties on ADME
ADME
Absorption
Distribution
Metabolism
Excretion
ADME for Oral administration
Gastrointestinal tract
Absorbed into circulatory, distributed
Absorbed into tissues
Can be metabolized
Excretion (from GI tract, circulatory systems, metabolic sites)
ADME for Intravenous injection
Circulatory systems
Absorbed by tissues
Metabolic sites
Excreted from GI tract, circulatory systems, tissues/metabolic sites
ADME for Intramuscular and Subcutaneous Injection
Tissues
Metabolic sites
absorbed into circulation
absorbed to GI tract
Excretion can happen at any point
Factors affecting drug absorption from GI
pharmaceutical
physicochemical
physiological
interactions
Pharmaceutical factors affecting drug absorption from GI
tablet
dissolutions/disintegration
excipients
Physiochemical factors affecting drug absorption from GI
solubility
hydrophilicity
lipophilicity
Ionization states
Physiological factors affecting drug absorption from GI
GI pH
metabolism
mucosal thickness (most absorption = thinnest = small intestine)
residence time (will spend most time in small intestine)
Interactions affecting drug absorption from GI
food
some foods must be taken on empty stomach
other drugs
antacids can decrease absorption
micro flora
milk/calcium products can interact with
Passive Absorption
energy independent
follows concentration gradient
dependent on ionization states
dependent on lipophilicity
dependent on duration and area of contact
drug molecule should be neutral (no charge)
Fick’s law of diffusion
need low thickness
high concentration difference
high area
high permeability
Factors affecting Passive Absorption
ionization states
lipophilicity
dissolution
interactions
Calculating % ionization of Acid
Calculating % ionization of a base
Ionization in GI tract
acidic drug: more ionized going down GI
basic drug: less ionized going down GI
Lipophilicity
affinity of a molecule or substance for lipid or aqueous environment
Dissolution
Solid dosage forms must dissolve into solution before absorption into blood
D= diffusion coefficient
A= surface area (of drug)
Cs= solubility of drug
Cb=concentration of drug in bulk
h=thickness of dissolution layer
Diffusion Coefficient (D)
value of D depends on particle size of molecule and viscosity of dissolution medium
presence of food in GI decreases D by increasing the viscosity of gastrointestinal fluids
Surface Area (A)
smaller particle size = greater surface area = higher dissolution rate
Solubility in diffusion layer (Cs)
directly proportional to intrinsic solubility
can be increased by salt formation
Particle structure (in dissolution)
polymorphism
different polymorphic forms have different solubility
amorphism
amorphous form dissolves more rapidly than crystalline form
may convert to crystalline form upon storage
Interactions
oral absorption of drugs may be affected by concurrent use of other substances/drugs that may
have large surface area upon which drug can be adsorbed (non-specific binding)
specifically bind/chelate drugs administered
alter gastric pH
antacids increase pH
alter gastrointestinal motility
higher motility = less absorption
lower motility = more absorption
Active Absorption
requires energy
movement against concentration gradient
saturable kinetics
selectively (in some cases)
competitive inhibition
drugs may act as substrates, inhibitors, or both
must have similar structure to natural substrates/inhibitors
Major transporters of active absorption
peptide transports
used by penicillin-based drugs
organic anion transporters
used by fexofenadine
organic cation transporters
used by metformin
Inhibitors of active absorption
grapefruit juice for organic anion transporter
Drug absorption in decreasing order
Solution > suspension > capsule > tablet > coated tablet
Drug role in dissolution/absorption
may be poorly soluble, hydrophobic
lubricant role in dissolution/absorption
usually hydrophobic
granulating agent role in dissolution/absorption
tends to hold ingredients together
filler role in dissolutoin/absorption
may interact with drug
should be water soluble
wetting agent role in dissolution/absorption
helps water penetration into tablet
disintegration agent role in dissolution/absorption
helps to break tablet apart
Volume of distribution (Vd)
fluid volume that would be required to accommodate the total amount of absorbed drug in the body at the plasma at steady state of concentration
amount of drug in body = drug absorbed (dose)
C = amount of drug in blood
not physical volume, extrapolated
drugs with low Vd stay within vascular compartment (blood)
bc of strong plasma protein binding or inability to diffuse out of circulation
drugs with high Vd are well-distributed throughout body or sequestered in tissue reservoirs (fatty tissues)
helpful in dose calculation and frequency of dosing
Factors Affecting rate of distribution
membrane permeability
blood perfusion
vital organs have high perfusion: lungs, kidneys, heart, liver
Factors affecting extent of distribution
lipid solubility
pH -pKa
Plasma protein binding
tissue drug binding
drugs bind specifically to certain tissue
ex. bisphosphonate (osteoporosis med) binds selectively to osteoclasts, stay bound longer so need less frequent dose
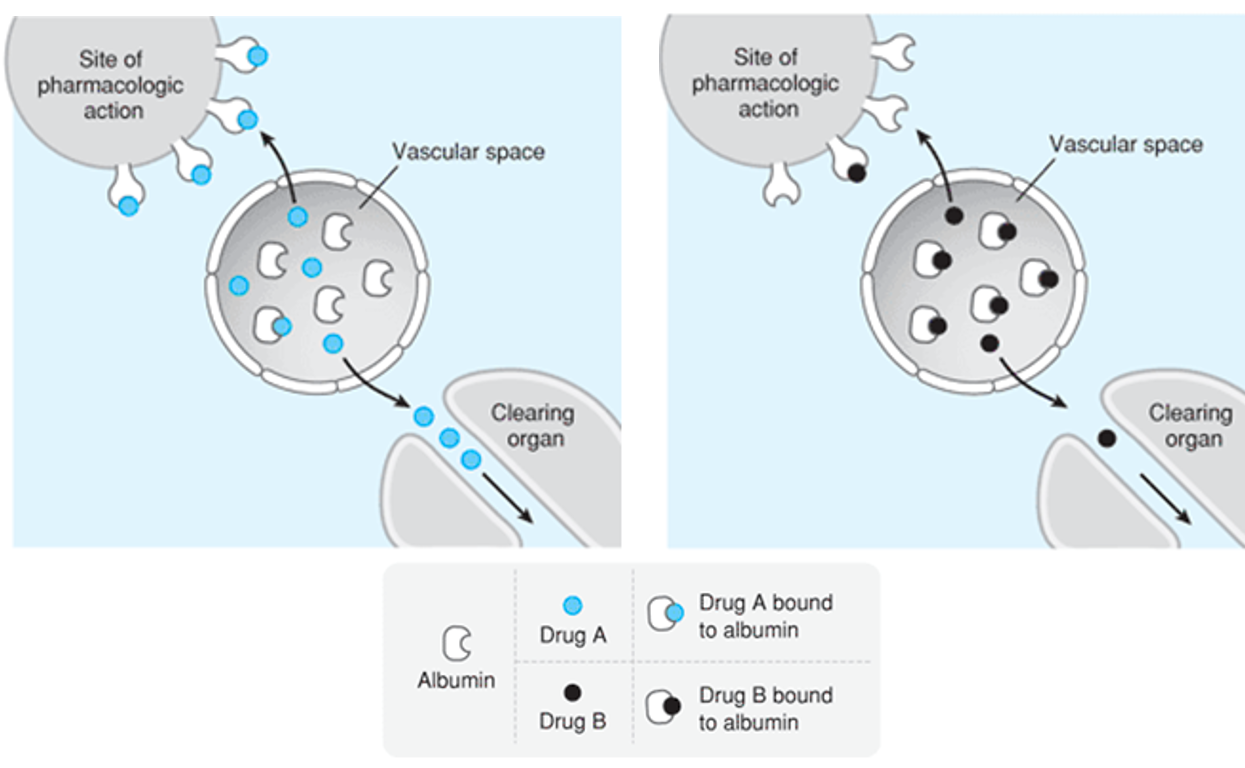
Plasma Protein Binding
drug must be free form to do biological action and be excreted
always equilibrium between bound and free forms
highly plasma protein bound drug = less free drug available to bind to site needed to perform action
Factors affecting Vd
lipophilicity
general rule:
higher lipophilicity = higher Vd
higher hydrophilicity = lower Vd
ionization
if drug is ionized at physiological pH, will not get distributed
stereochemistry
physiological
any disease that changes blood volume
age and gender
skeletal muscles decrease with age
children have different Vd vs adults
hormonal diff between genders can affect
Drug metabolism
the process by which the body chemically alters drugs to make them easier to excrete and reduce their toxicity
increase water solubility
2 phases
not all drugs can undergo
Factors affecting Metabolism
inhibitors of metabolizing enzymes
inducers of metabolizing enzymes
age and gender
stereochemistry
genetic variation
physiological factors
Excretion
irreversible loss of drug from body
primarily via hepatic and renal route
renal (urination) more common, preferred
must be water-soluble
also feces, sweat, saliva, tears, exhaled air (ex. alcohol), breast milk
Stages in renal excretion
Passive glomerular filtration
depends on physiochemical properties
molecules must be neutral
Active tubular secretion
Passive tubular re-absorption
Renal Clearance
rate of excretion/plasma conc
Factors Affecting Renal Excretion
age
with age, renal function declines
gender
hormonal differences
renal dysfunction
drugs inhibiting active secretion (probenecid)
Probenecid
gout medicine
in past, was given in combination with penicillin to inhibit active tubular secretion (so it stayed in body longer)
Enteral routes of administration
oral
parenteral routes of administration
intravenous
subcutaneous
intramuscular
intrathecal
intraperitoneal
Mucosal routes of administration
sublingual
ocular
nasal
pulmonary
rectal
urinary
reproductive tract
topical routes of administration
dermal
Oral dosage forms
tablets
capsules
suspensions
solutions
syrups
emulsions
Oral dosage form requirements
stable in GI tract (chemical and metabolic stability)
stable at physiological pH of GI tract
stable against stomach enzymes
absorbable (passive/active transport)
Oral dosage form advantages
simple
inexpensive
convenient
less invasive
oral dosage form disadvantages
degradation of drug in GI tract
first-pass metabolism
slow onset of action
cannot use in emergency
First pass metabolism
metabolism happens before drug gets into circulation (in liver, GI tract)
drug metabolism at a specific location in the body which leads to a reduction in the concentration of the active drug before it reaches the site of action or systemic circulation.
intramuscular route of admin
parenteral
absorption:
prompt absorption from aqueous solutions
slow absorption from repository depot preparations
75-100% bioavailability
can be used with moderate volumes, oily vehicles, some irritants, depot (sustained-release) injections
Limitations
cannot be used concurrently with anti-coagulant therapy
vasular damage/bruising
may interfere with certain diagnostic tests
Intrathecal route of admin
parenteral, injection into spine
very fast absorption
provides fast and effective spinal block of pain perception
allows for maximizing drug concentration in CNS
limited volume
issues relating to dural puncture (infection, loss of pressure/fluid, headaches, dizziness)
ex. Epidural pain relief, anticancer agents for brain cancer
Intravenous/Intraarterial route of drug admin
parenteral
Absorption:
circumvents need for cross-membrane absorption to get drug into circulation
effects may be immediate or almost immediate (good or bad)
100% bioavailability
Special utility
best route for emergency use
can be used with larger volumes/irritants
can be used for larger peptides and proteins
Limitations
not suitable for poorly soluble or insoluble agents, oily vehicles
most cases require slow administration
increased risk of adverse events
Subcutaneous route of admin
parenteral
absorption:
prompt (aqueous)
slow (repository prep)
75-100% bioavailability
special utility
could be used for relatively insoluble suspensions
limitations
limited volumes
irritation at injection site
pain
tissue necrosis
Buccal/Sublingual route of drug admin
mucosal
not oral: remain in mouth, does not go to GI
very prompt absorption
60-80% bioavailability
best route for treatment of periodontal disease
allows for avoidance of first-pass effect (“facial triangle”)
limitation: swallowing medication
if swallowed, will not work (dose is smaller than oral)
Intranasal route of admin
mucosal
prompt absorption
best route for treatment of nasopharyngeal diseases (nasal congestion, sinus infection)
limitations
limited volumes
irritation
probable systemic effects
Ocular route of admin
mucosal
prompt absorption
best route for treatment of ophthalmic disease
limitations
limited volumes
irritation
possible systemic effects
Pulmonary/Inhalation route of admin
mucosal
prompt absorption
5-100% bioavailability
best route for treatment of pulmonary disease
limitations
limited volumes
possible irritation
probable systemic effects
Rectal route of admin
mucosal
prompt absorption
30-100% bioavailability
best route for treatment of anorectal diseases (hemorrhoids)
optional route when oral delivery not feasible (vomiting, unconscious)
avoids most of first-pass effect
limitations:
limited volumtes
irritation
probable systemic effects
absorption can be irregular/incomplete
diarrhea
Epidermal/Transdermal route of admin
topical
variable absorption if skin abraded or inflamed
best absorption with lipid-soluble drugs (steroids)
80-100% bioavailability
best option for treatment of skin diseases
convenient (transdermals)
limitations
limited volumes
possible irritation
variable absorption
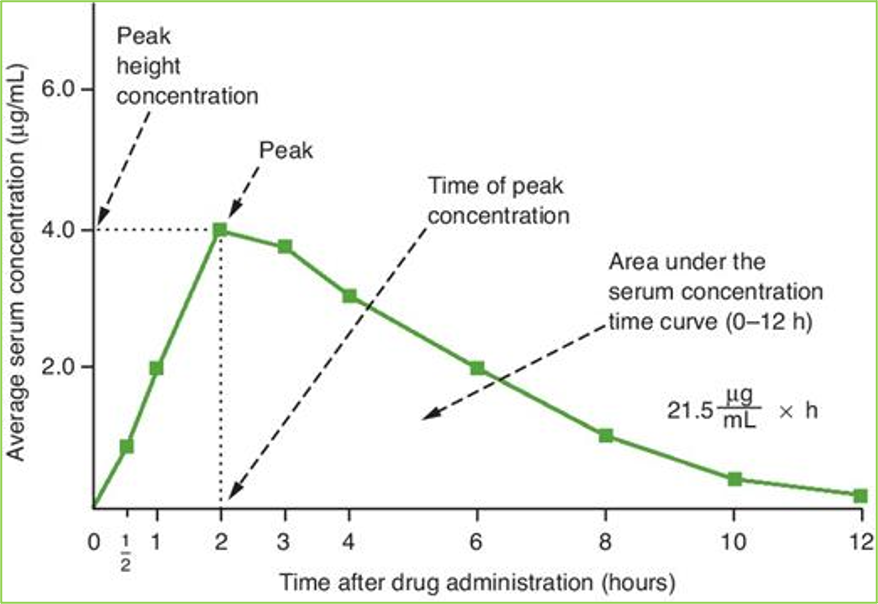
Bioavailability
rate and extent to which an active drug ingredient or therapeutic moiety is absorbed from a drug product and becomes available at the site of action
graphically, conc vs time
Peak: highest drug conc in blood serum
Peak time: time it takes to reahc peak
area under curve compared to compare bioavailabilities
Bioavailability utility
determine amount/proportion of drug absorbed from dosage form
determine rate of drug absorption
determine duration of drug presence in biologic fluid/tissue
determine relationship between drug blood levels and clinical effectiveness/toxicity
Factors affecting bioavailability
ADME profile of drug
enterohepatic circulation
drug absorbed, gets into liver, liver throws back to small intestine, which moves it back to liver
presystemic metabolism
“first pass” metabolism
oral drug can get paralyzed in GI, GI wall, liver before absorbed into circulation
Bioavailability formula
max =100%
IV route
Minimum effective concentration
conc of drug required to produce effect
reaches conc at onset time
Duration of action
how long drug stays above/at minimum effective concentration
Relative bioavailability
bioavailabilites of same drug in two different formulations
Bioequivalence
pharmaceutical alternatives or equivalents which have exact same bioavailabilities when administered at same dose
bioequivalence studies performed to compare extent of absorption of new product or generic products
have same bioavailibility curve (same onset, peak, min effective concentration)
Methods to assess bioavailability
dissolution at administration or absorption site
evaluates dissolution rate
Free drug in systemic circulations
evaluates:
blood level time profile
peak blood level
time to reach peak
area under blood level time curve
Pharmacologic effect
evaluates:
onset of effect
duration of effect
intensity of effect
Clinical response
controlled clinical blind or double-blind study, observed clinical success or failure
Factors affecting dosage regimen
activity, toxicity
pharmacokinetics
clinical state
management of therapy
other factors
Activity/Toxicity and dosage considerations
minimum therapeutic dose
toxic dose
therapeutic index
diff btwn conc of drug between minimum toxic conc (largest conc before toxic) and minimum effective conc
larger range = wide index
narrow index: calculate dose carefully
side-effects
dose-response relationships
Pharmacokinetics dosage considerations
ADME
clinical state dosage considerations
age, weight, urine pH
urine pH crucial for excretion
condition being treated
existence of other disease
Management of therapy and dosage considerations
multiple drugs
convenience of regimen
patient compliance
other factors in dosage considerations
tolerance -dependence
pharmacogenomics
drug interactions
lifestyle factors